Воспроизведение полнотекстовой диаграммы статьи об одноклеточном анализе данных: истощение NK-клеток и траектория эволюции опухолевых клеток
Обзор статьи
Название статьи: «Комплексный транскриптомный и протеомный анализ отдельных клеток выявляет истощение NK-клеток и уникальную траекторию эволюции опухолевых клеток при неороговевающей карциноме носоглотки».
Дата публикации и журнал: Опубликовано в журнале трансляционной медицины в 2023 г.
Ссылка для онлайн-чтения: https://doi.org/10.1186%2Fs12967-023-04112-8
Статья Экспериментальный дизайн
основнойИзучить роль NK-клеток и опухолевых клеток в неороговевающем раке носоглотки (NK-NPC), который тесно связан с инфекцией EBV.
метод:Три образца NK-NPC и три образца нормальной слизистой оболочки носоглотки были собраны для анализа протеома и проанализированы в сочетании с GSE162025 (данные одноклеточного транскриптома NK-NPC) и GSE150825 (данные одноклеточного транскриптома NLH).
Этапы анализа:
- Анализ данных протеома,Было обнаружено, что белки, опосредованные цитотоксичностью, опосредованные NK-клетками, значительно снижаются.,Функция NK-клеток может быть подавлена.
- Иммуногистохимия показала, что уровни B2M, CD56+ и гранзима B, связанные с NK-клетками, были снижены в образцах NK-NPC.
- Анализ образцов транскриптома отдельных клеток NK-NPC показал, что Т-клетки, В-клетки и NK-клетки входят в тройку лучших среди всех типов клеток.
- Возьмите подмножество NK-клеток и разделите их, чтобы получить 1 мачту. ячейка3 кластер NK-клеток,Активация/ингибирование цитотоксичности NK,сотовая связь,Маркеры, связанные с функцией NK-клеток,Анализ с точки зрения путей токсичности, опосредованной NK-клетками,и подтверждено с использованием набора одноклеточных транскриптомов NLH
- Подмножество эпителиальных клеток было разделено и использовано copykat для идентификации доброкачественных и злокачественных эпителиальных клеток. Злокачественные эпителиальные клетки были дополнительно подразделены для получения категорий опухоль1 и опухоль2. Было исследовано множество ключевых генов, и было обнаружено, что инфекция EBV может быть связана с инфекцией EBV. опухоль1.
- Затем выделили опухоль1, чтобы получить четыре кластера. Мы использовали квазихронологический анализ для анализа процесса эволюции между четырьмя кластерами. Мы проанализировали изменения в ключевых генах и путях между различными кластерами и предположили механизм заражения ВЭБ.
Процесс воспроизводства
Загрузите необходимые пакеты R:
library(Seurat)
library(data.table)
library(stringr)
library(readxl)
library(tibble)
library(limma)
library(dplyr)
library(ggplot2)
library(ggrepel)
library(pheatmap)
library(clusterProfiler)
library(org.Hs.eg.db)
library(tidyverse)
library(scRNAtoolVis)
library(enrichplot)
library(GSEABase)
library(msigdbr)
library(CellChat)
library(copykat)
library(patchwork)
library(SCORPIUS)
Данные по протеому можно скачать во вложении. Нормализованная численность представляет собой количественный результат, а лимма используется для анализа различий.
# proteomics
data_pro <- read_xlsx('../12967_2023_4112_MOESM1_ESM.xlsx')
data_pro <- data_pro[,c(23,37:42)]
colnames(data_pro) <- c('gene symbol','NK_NPC-1','NK_NPC-2','NK_NPC-3','normal-1','normal-2','normal-3')
data_pro$`gene symbol` <- str_split(data_pro$`gene symbol`,';',simplify = T)[,1]
data_pro <- data_pro[!duplicated(data_pro$`gene symbol`),]
data_pro <- data_pro[!is.na(data_pro$`gene symbol`),]
data_pro <- column_to_rownames(data_pro,'gene symbol')
data_pro <- data_pro[rowSums(!is.na(data_pro))==6,]
# deg
input_matrix <- log2(data_pro)
group <- as.factor(rep(c('NK_NPC','normal'),each=3))
design <- model.matrix(~0+group)
colnames(design) <- c('NK_NPC','normal')
rownames(design) <- colnames(input_matrix)
fit <- lmFit(input_matrix,design)
con.matrix <- makeContrasts('NK_NPC-normal',levels = design)
fit2 <- contrasts.fit(fit,contrasts = con.matrix)
fit2 <- eBayes(fit2)
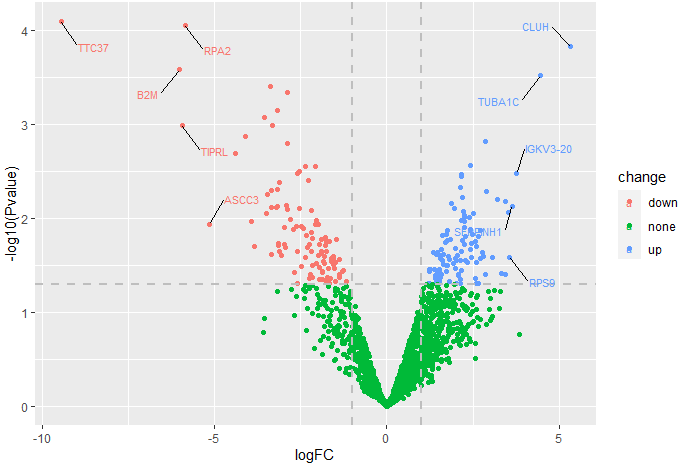
# дифференциальная фильтрация
options(digits = 4)
deg <- topTable(fit2,n=Inf)
up <- (deg$logFC>0.5)&(deg$P.Val<0.05)
down <- (deg$logFC<(-0.5))&(deg$P.Val<0.05)
change <- ifelse(up,'up',ifelse(down,'down','none'))
deg <- mutate(deg,change)
deg <- mutate(deg,'p' = -log10(deg$P.Val))
top5 <- rownames(top_n(deg[deg$change=='up',],5,logFC))
down5 <- rownames(top_n(deg[deg$change=='down',],-5,logFC))
deg$label <- ifelse(rownames(deg) %in% c(top5,down5),rownames(deg),NA)
# volcano
ggplot(deg,aes(x=logFC,y=p,color=change))+
geom_point()+
geom_vline(xintercept = 1,linetype=2,color='gray',linewidth=1)+
geom_vline(xintercept = -1,linetype=2,color='gray',linewidth=1)+
geom_hline(yintercept = -log10(0.05),linetype=2,color='gray',linewidth=1)+
geom_text_repel(aes(logFC,label=label),
max.overlaps = 100000,size=3,segment.color='black',
box.padding=unit(1,'lines'))
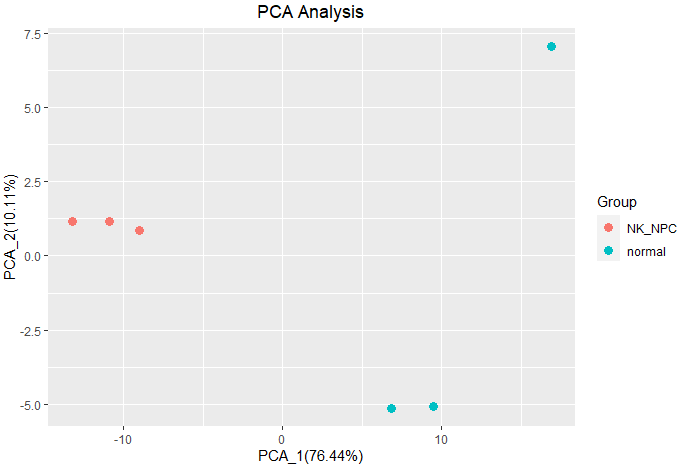
# дифференциальный белок PCA
pca_data <- data_pro[rownames(deg[deg$change!='none',]),]
pca <- prcomp(t(log2(pca_data)),scale. = T)
xlab <- paste0('PCA_1(', round(summary(pca)$importance[2,1]*100,2),'%)')
ylab <- paste0('PCA_2(', round(summary(pca)$importance[2,2]*100,2),'%)')
ggplot(data.frame(pca$x),aes(x=PC1,y=PC2,color=group))+
geom_point(size = 3)+stat_ellipse(level = 0.95, show.legend = F)+
labs(x = xlab,y = ylab,title = 'PCA Analysis')+
theme(plot.title = element_text(hjust = 0.5))+
guides(color = guide_legend(title = 'Group'))
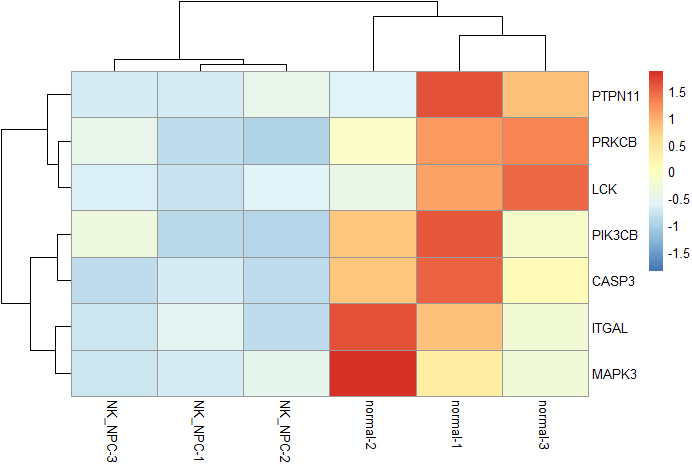
# heatmap
NK_cytotoxicity <- c('PRKCB','PTPN11','LCK','ITGAL','PIK3CB','MAPK3','CASP3')
pheatmap(data_pro[NK_cytotoxicity,],scale = 'row')
# KEGG
kegg_id <- bitr(rownames(deg[deg$change!='none',]),fromType = 'SYMBOL',toType = 'ENTREZID',OrgDb = org.Hs.eg.db)
kegg <- enrichKEGG(kegg_id$ENTREZID,'hsa',keyType = 'kegg')
kegg <- setReadable(kegg,OrgDb = org.Hs.eg.db,keyType = 'ENTREZID')
fc <- deg[deg$change!='none','logFC']
names(fc) <- rownames(deg[deg$change!='none',])
cnetplot(kegg,color.params = list(foldChange = fc,edge=T))
Результаты анализа различий:

Анализ данных отдельных клеток NK-NPC
Затем проанализируйте данные одной ячейки NK-NPC.
Чтение данных
# Чтение данных
# 162025
data_162025 <- fread('./GSE162025_RAW/Tumor/GSM4929846_NPC_SC_1802_Tumor_count.csv.gz')
filename <- paste('GSE162025_RAW/Tumor/',list.files('GSE162025_RAW/Tumor/'),sep = '')
data_162025List <- lapply(filename, function(x){
obj <- CreateSeuratObject(counts = read.csv(x),
# project = paste(str_split(str_split(x,'/')[[1]][3],'_')[[1]][2:4],collapse = '_'),
min.cells = 3, min.features = 200)
})
data_162025 <- merge(data_162025List[[1]],data_162025List[-1],add.cell.ids = names(data_162025List))
data_162025$orig.ident <- apply(str_split(rownames(data_162025@meta.data),'_',simplify = T)[,1:3],
1,
function(x){paste0(x,collapse ='_')})
dim(data_162025)
saveRDS(data_162025,file = 'data_162025.RDS')
data_162025 <- readRDS('data_162025.RDS')
Последующий процесс анализа: контроль качества, кластеризация с уменьшением размерности и аннотация.
# QC
VlnPlot(data_162025,features = 'nFeature_RNA',group.by = 'orig.ident',pt.size = 0)
data_162025 <- subset(data_162025,subset = nFeature_RNA>200 & nFeature_RNA<4000)
data_162025 <- PercentageFeatureSet(data_162025,pattern = '^MT',col.name = 'percent.MT')
VlnPlot(data_162025,features = 'percent.MT',group.by = 'orig.ident',pt.size = 0)
data_162025 <- subset(data_162025,subset = percent.MT<5)
dim(data_162025)
# Уменьшение размерности
data_162025 <- NormalizeData(data_162025)
data_162025 <- FindVariableFeatures(data_162025)
data_162025 <- ScaleData(data_162025)
data_162025 <- RunPCA(data_162025)
ElbowPlot(data_162025,ndims = 30)
# Группировка
data_162025 <- RunTSNE(data_162025,reduction = 'pca',dims = 1:20)
DimPlot(data_162025,reduction = 'tsne',label = T,group.by = 'orig.ident')
data_162025 <- FindNeighbors(data_162025,dims = 1:20)
data_162025 <- FindClusters(data_162025,resolution = 0.8)
DimPlot(data_162025,reduction = 'tsne',label = T,group.by = 'seurat_clusters')
# Комментарий
# T cell ['PTPRC','CD3D','CD8A']---[0,1,3,4,5,7,8,9,10,11,12,13]
# NK cell ['PTPRC','NKG7','CD96','KLRC1','NCAM1']---[7,11] [0,1,5,7,10,11,13]
# Myeloid cells ['CD14', 'CD68', 'CD163', 'ITGAX','FCGR3A']---[15,18,19]
# Epithelial cells ['EPCAM', 'KRT5', 'TP63', 'SSTR2']---[16]
# B cell ['CD19', 'MS4A1', 'CD79A']---[2,6,14,17]
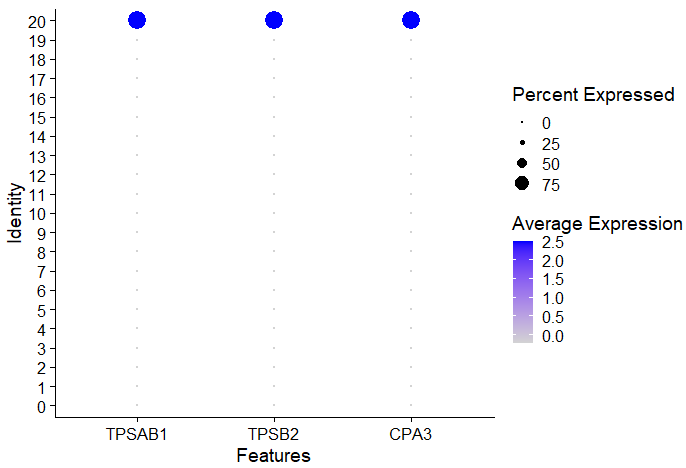
# mast cell ['TPSAB1','TPSB2','CPA3'] --- [20]
cell_type <- data.frame(cluster = 0:20,type = 0:20)
cell_type[cell_type$cluster %in% c(0,1,3,4,5,7,8,9,10,11,12,13),'type'] <- 'T cells'
cell_type[cell_type$cluster %in% c(7,11,20),'type'] <- 'NK cell'
cell_type[cell_type$cluster %in% c(15,18,19),'type'] <- 'Myeloid cells'
cell_type[cell_type$cluster %in% c(16),'type'] <- 'Epithelial cells'
cell_type[cell_type$cluster %in% c(2,6,14,17),'type'] <- 'B cells'
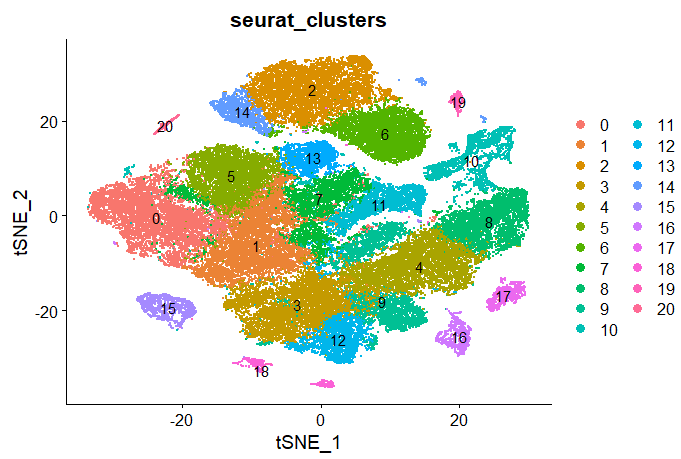
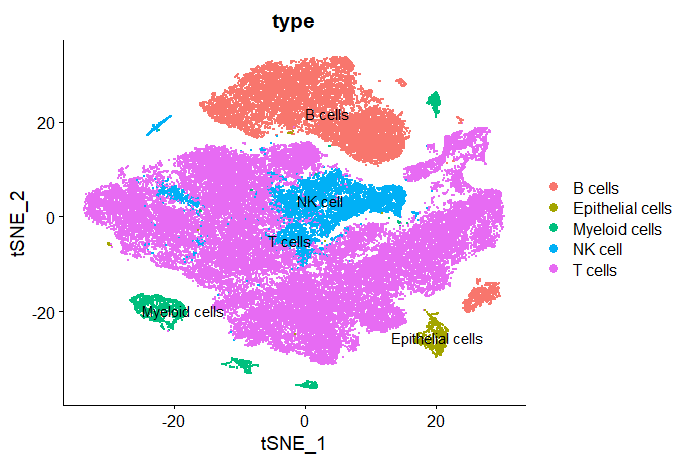
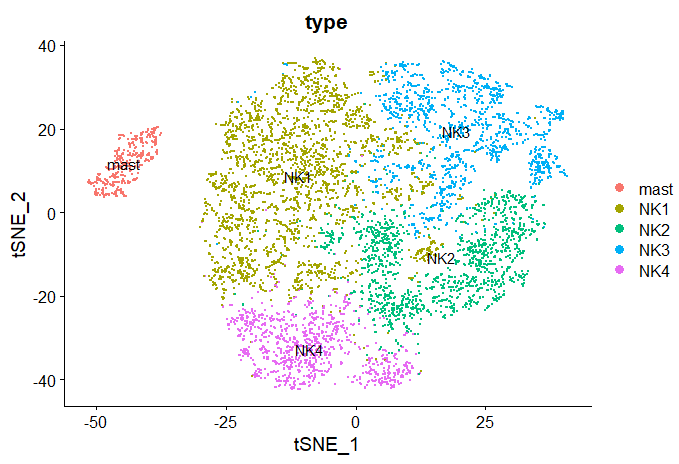
Результат на самом деле очень очевиден. Cluster20 — это тучная клетка, и ее можно напрямую отделить. Я не знаю, почему ее следует отнести к NK-клетке, а затем подразделить.
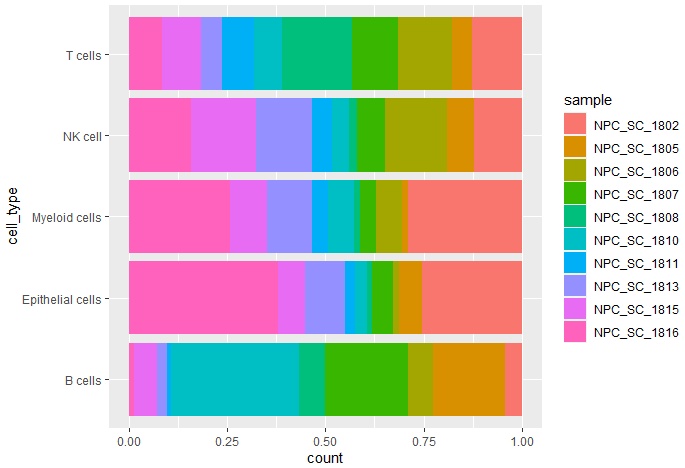
# Составная гистограмма пропорций типов клеток
cell_ratio <- matrix(nrow = length(unique(data_162025@meta.data$type)),
ncol = length(unique(data_162025@meta.data$orig.ident)))
cell_ratio <- as.data.frame(cell_ratio,row.names = unique(data_162025@meta.data$type))
colnames(cell_ratio) <- unique(data_162025@meta.data$orig.ident)
table(data_162025@meta.data$orig.ident)
for (cell in unique(data_162025@meta.data$type)){
cell_count <- sum(data_162025@meta.data$type==cell)
for (sample in unique(data_162025@meta.data$orig.ident)){
cell_ratio[cell,sample] <- sum(data_162025@meta.data$type==cell & data_162025@meta.data$orig.ident==sample) / cell_count
}
}
cell_ratio <- rownames_to_column(cell_ratio,var = 'cell_type')
cell_ratio <- pivot_longer(cell_ratio,cols = -cell_type,names_to = 'sample')
ggplot(cell_ratio, aes( x = cell_type, weight = value, fill = sample))+
geom_bar( position = "stack")+coord_flip()
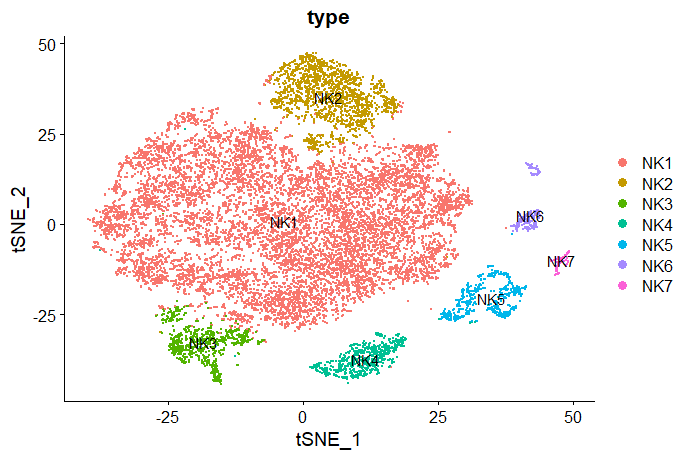
Продолжайте подразделять NK-клетки
NK_subtype <- subset(data_162025,subset= type == 'NK cell')
NK_subtype <- NormalizeData(NK_subtype)
NK_subtype <- FindVariableFeatures(NK_subtype)
NK_subtype <- ScaleData(NK_subtype)
NK_subtype <- RunPCA(NK_subtype)
ElbowPlot(NK_subtype,ndims = 30)
NK_subtype <- RunTSNE(NK_subtype,reduction = 'pca',dims = 1:10)
DimPlot(NK_subtype,reduction = 'tsne')
NK_subtype <- FindNeighbors(NK_subtype,dims = 1:10)
NK_subtype <- FindClusters(NK_subtype,resolution = 0.15)
DimPlot(NK_subtype,reduction = 'tsne',group.by = 'seurat_clusters',label = T)
NK_subtype$type <- ifelse(NK_subtype$seurat_clusters=='0','NK1',
ifelse(NK_subtype$seurat_clusters=='1','NK2',
ifelse(NK_subtype$seurat_clusters=='2','NK3',
ifelse(NK_subtype$seurat_clusters=='3','NK4','mast'))))
DimPlot(NK_subtype,reduction = 'tsne',group.by = 'type',label = T)
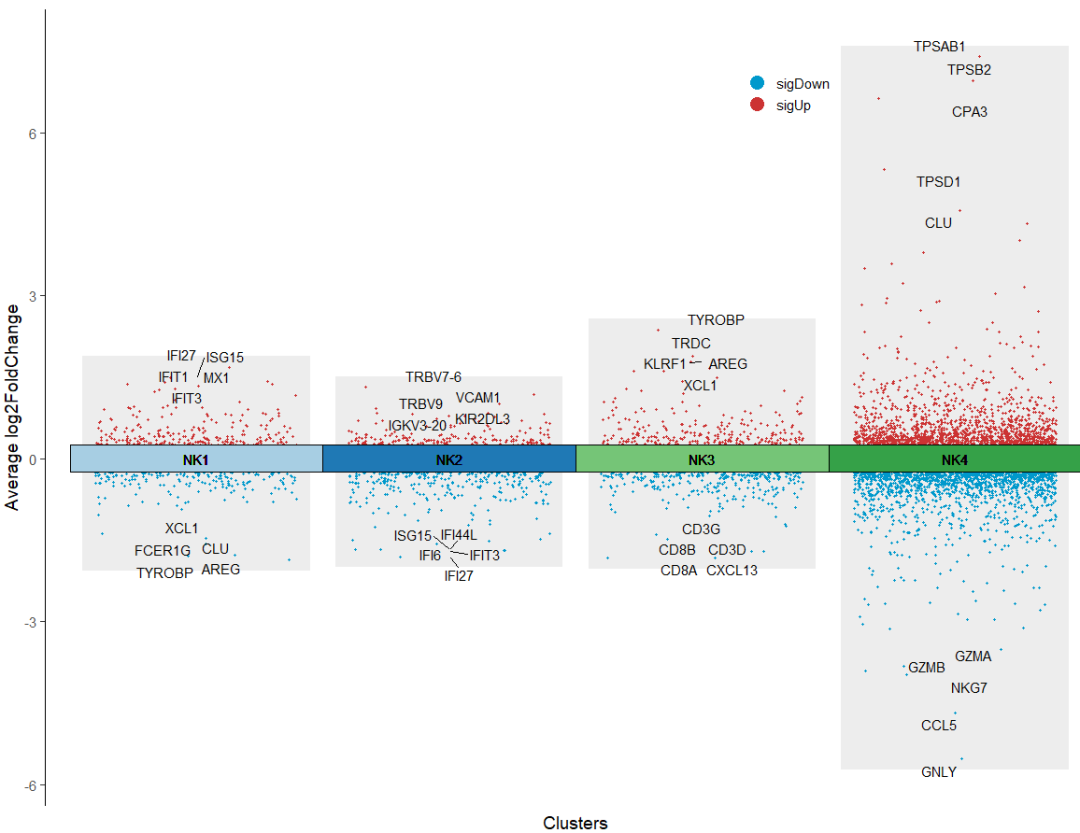
# scRNA volcano
NK_deg <- FindAllMarkers(NK_subtype)
NK_deg$cluster <- ifelse(NK_deg$cluster=='0','NK1',
ifelse(NK_deg$cluster=='1','NK2',
ifelse(NK_deg$cluster=='2','NK3','NK4')))
jjVolcano(NK_deg)
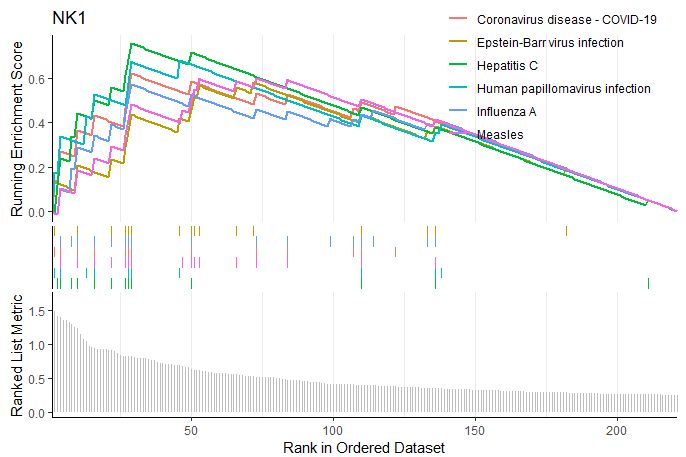
Анализ GSEA
соответственно4индивидуальныйNKклеткасубпопуляциярегулирование вверх и внизgeneруководить Анализ GSEA
# GSEA
NK_1df <- NK_deg[NK_deg$cluster=='NK1',]
NK_1_up <- NK_1df[NK_1df$avg_log2FC>0.25 & NK_1df$p_val_adj<0.05,'avg_log2FC']
names(NK_1_up) <- bitr(NK_1df[NK_1df$avg_log2FC>0.25 & NK_1df$p_val_adj<0.05,'gene'],
fromType = 'SYMBOL',toType = 'ENTREZID',OrgDb = org.Hs.eg.db)[,'ENTREZID']
NK_1_up <- sort(NK_1_up,decreasing = T)
NK_1_up_gse <- gseKEGG(NK_1_up)
dim(NK_1_up_gse@result)
NK_1_down <- NK_1df[NK_1df$avg_log2FC<(-0.25) & NK_1df$p_val_adj<0.05,'avg_log2FC']
names(NK_1_down) <- bitr(NK_1df[NK_1df$avg_log2FC<(-0.25) & NK_1df$p_val_adj<0.05,'gene'],
fromType = 'SYMBOL',toType = 'ENTREZID',OrgDb = org.Hs.eg.db)[,'ENTREZID']
NK_1_down <- sort(NK_1_down,decreasing = T)
NK_1_down_gse <- gseKEGG(NK_1_down)
dim(NK_1_down_gse@result)
gseaplot2(NK_1_up_gse,geneSetID = 1:6,title = 'NK1')
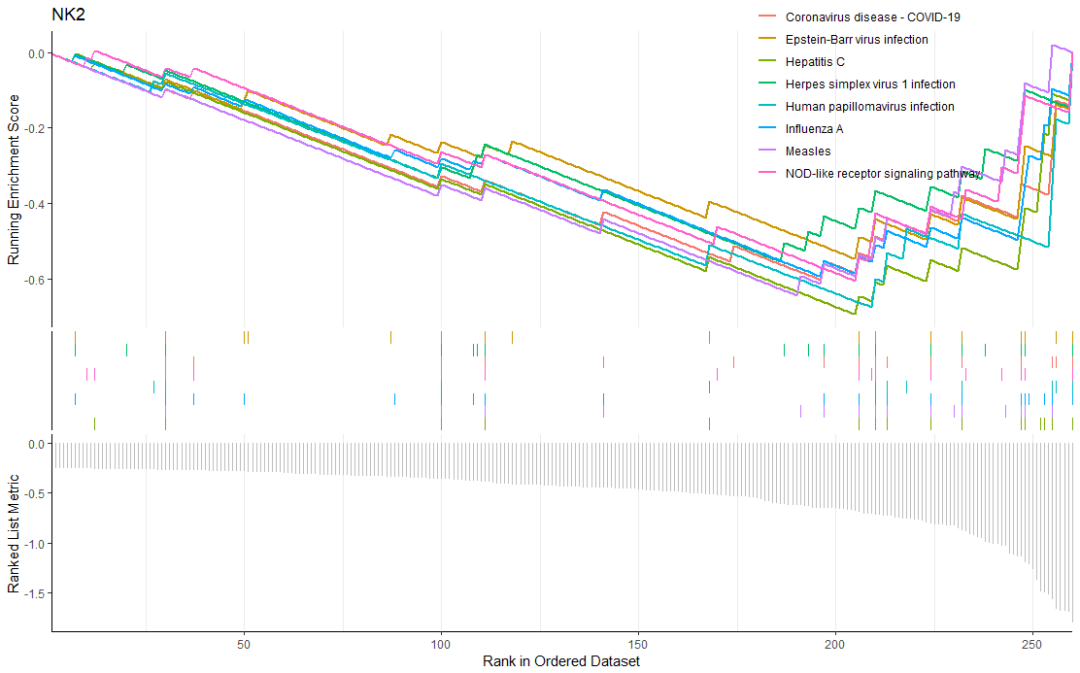
############ NK2
NK_2df <- NK_deg[NK_deg$cluster=='NK2',]
NK_2_up <- NK_2df[NK_2df$avg_log2FC>0.25 & NK_2df$p_val_adj<0.05,'avg_log2FC']
names(NK_2_up) <- bitr(NK_2df[NK_2df$avg_log2FC>0.25 & NK_2df$p_val_adj<0.05,'gene'],
fromType = 'SYMBOL',toType = 'ENTREZID',OrgDb = org.Hs.eg.db)[,'ENTREZID']
NK_2_up <- sort(NK_2_up,decreasing = T)
NK_2_up_gse <- gseKEGG(NK_2_up)
NK_2_down <- NK_2df[NK_2df$avg_log2FC<(-0.25) & NK_2df$p_val_adj<0.05,'avg_log2FC']
names(NK_2_down) <- bitr(NK_2df[NK_2df$avg_log2FC<(-0.25) & NK_2df$p_val_adj<0.05,'gene'],
fromType = 'SYMBOL',toType = 'ENTREZID',OrgDb = org.Hs.eg.db)[,'ENTREZID']
NK_2_down <- sort(NK_2_down,decreasing = T)
NK_2_down_gse <- gseKEGG(NK_2_down)
dim(NK_2_down_gse@result)
gseaplot2(NK_2_down_gse,geneSetID = 1:8,title = 'NK2')
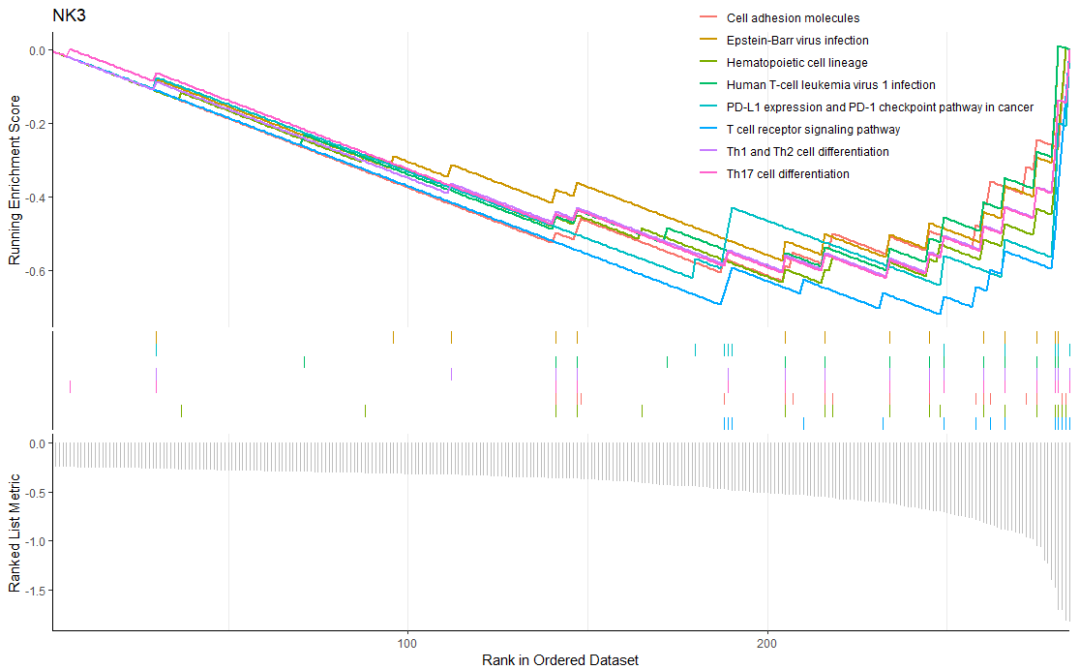
############ NK3
NK_3df <- NK_deg[NK_deg$cluster=='NK3',]
NK_3_up <- NK_3df[NK_3df$avg_log2FC>0.25 & NK_3df$p_val_adj<0.05,'avg_log2FC']
names(NK_3_up) <- bitr(NK_3df[NK_3df$avg_log2FC>0.25 & NK_3df$p_val_adj<0.05,'gene'],
fromType = 'SYMBOL',toType = 'ENTREZID',OrgDb = org.Hs.eg.db)[,'ENTREZID']
NK_3_up <- sort(NK_3_up,decreasing = T)
NK_3_up_gse <- gseKEGG(NK_3_up)
dim(NK_3_up_gse@result)
NK_3_down <- NK_3df[NK_3df$avg_log2FC<(-0.25) & NK_3df$p_val_adj<0.05,'avg_log2FC']
names(NK_3_down) <- bitr(NK_3df[NK_3df$avg_log2FC<(-0.25) & NK_3df$p_val_adj<0.05,'gene'],
fromType = 'SYMBOL',toType = 'ENTREZID',OrgDb = org.Hs.eg.db)[,'ENTREZID']
NK_3_down <- sort(NK_3_down,decreasing = T)
NK_3_down_gse <- gseKEGG(NK_3_down)
dim(NK_3_down_gse@result)
gseaplot2(NK_3_up_gse,geneSetID = 1,title = NK_3_up_gse@result$Description)
gseaplot2(NK_3_down_gse,geneSetID = 1:8,title = 'NK3')
############ NK4
NK_4df <- NK_deg[NK_deg$cluster=='NK4',]
NK_4_up <- NK_4df[NK_4df$avg_log2FC>0.25 & NK_4df$p_val_adj<0.05,'avg_log2FC']
names(NK_4_up) <- bitr(NK_4df[NK_4df$avg_log2FC>0.25 & NK_4df$p_val_adj<0.05,'gene'],
fromType = 'SYMBOL',toType = 'ENTREZID',OrgDb = org.Hs.eg.db)[,'ENTREZID']
NK_4_up <- sort(NK_4_up,decreasing = T)
NK_4_up_gse <- gseKEGG(NK_4_up)
dim(NK_4_up_gse@result)
NK_4_down <- NK_4df[NK_4df$avg_log2FC<(-0.25) & NK_4df$p_val_adj<0.05,'avg_log2FC']
names(NK_4_down) <- bitr(NK_4df[NK_4df$avg_log2FC<(-0.25) & NK_4df$p_val_adj<0.05,'gene'],
fromType = 'SYMBOL',toType = 'ENTREZID',OrgDb = org.Hs.eg.db)[,'ENTREZID']
NK_4_down <- sort(NK_4_down,decreasing = T)
NK_4_down_gse <- gseKEGG(NK_4_down)
dim(NK_4_down_gse@result)
gseaplot2(NK_4_down_gse,geneSetID = 1:4)
NK1
NK2
NK3
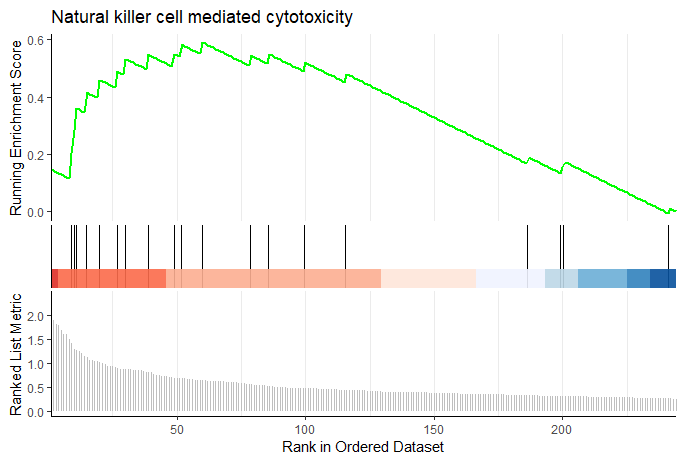
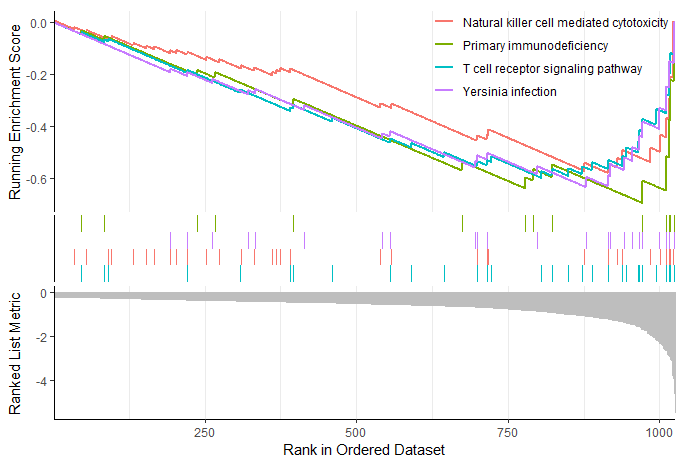
Видно, что цитотоксичность, опосредованная субпопуляцией NK3, активируется, а цитотоксичность, опосредованная субпопуляцией NK4, ингибируется.
Анализ сотовой связи
# cellchat
# Шаг 1. Создайте объект сотового чата.
data.input <- NK_subtype@assays$RNA@data
meta.data <- NK_subtype@meta.data
cellchat <- createCellChat(object=data.input,
meta = meta.data,
group.by='type')
cellchat <- addMeta(cellchat,meta = meta.data)
# Шаг2. Загрузите библиотеку CellChatDB.
cellchatDB <- CellChatDB.human
cellchat@DB <- cellchatDB
# Шаг 3. Обработайте данные выражения.
cellchat <- subsetData(cellchat)
cellchat <- identifyOverExpressedGenes(cellchat)
cellchat <- identifyOverExpressedInteractions(cellchat)
# Шаг 4. Рассчитайте вероятность связи и сделайте вывод о сети сотовой связи.
cellchat <- computeCommunProb(cellchat,population.size = F)
cellchat <- filterCommunication(cellchat,min.cells = 10)
# Step5. Извлеките прогнозируемую сеть сотовой связи в виде данных. frame
df.net <- subsetCommunication(cellchat)
df.pathway <- subsetCommunication(cellchat,slot.name = 'netP')
# Step6. Вывод о клеточной связи на уровне сигнального пути
cellchat <- computeCommunProbPathway(cellchat)
# Step7. Вычисление и добавление сети межклеточной связи
cellchat <- aggregateNet(cellchat)
saveRDS(cellchat,file = 'cellchat.RDS')
levels(cellchat@idents)
netVisual_bubble(cellchat, sources.use = c(1),
targets.use = c(2,3,4,5),remove.isolate = FALSE)
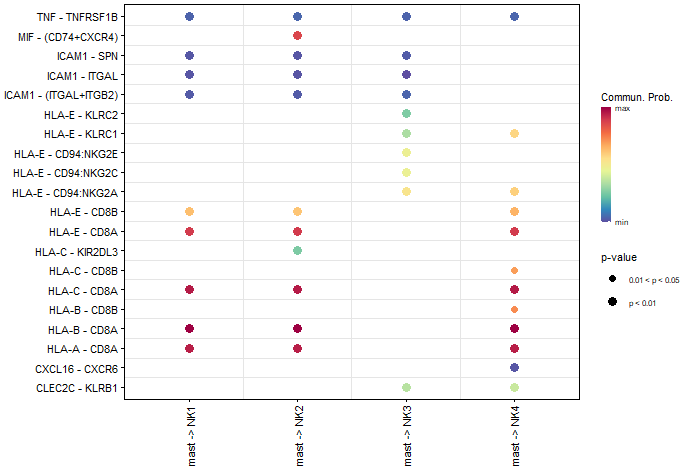
Вы можете видеть взаимодействие между тучными клетками и подтипом NK, но сигнал ГАЛЕКТИН не кажется значимым.
Визуализация маркеров клеточного истощения в NK-клетках
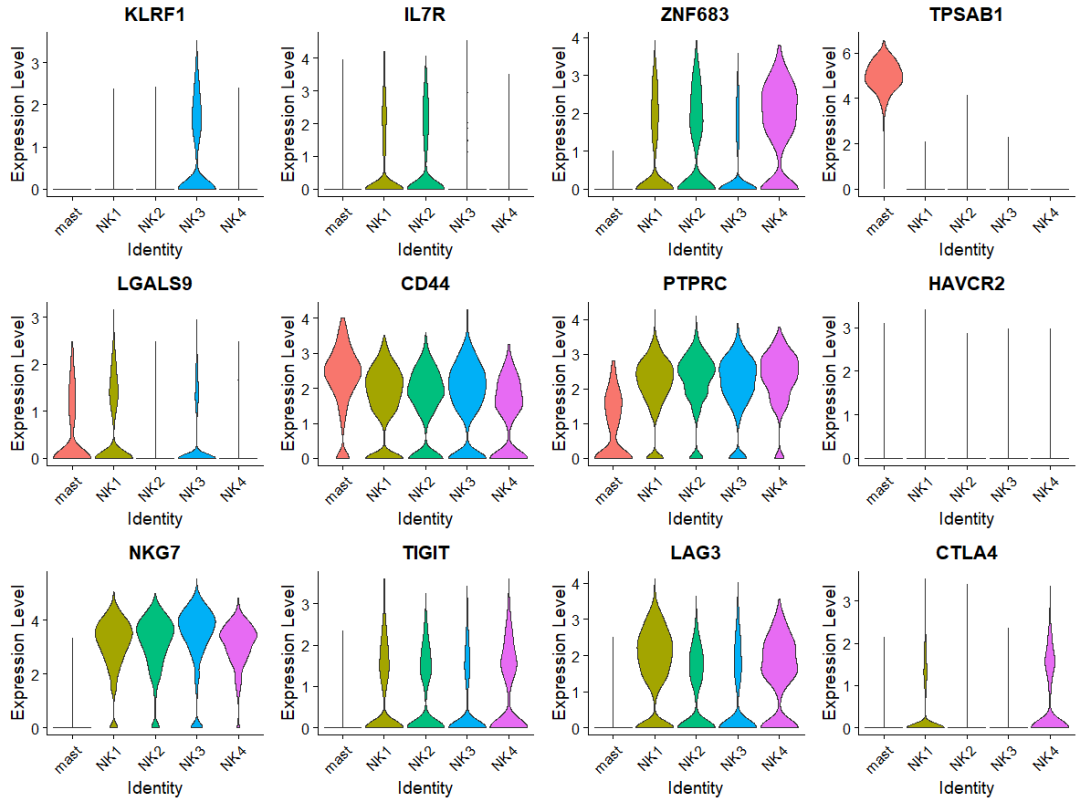
Глядя на экспрессию маркеров клеточного истощения HAVCR2, TIGIT, LAG3 и CTLA4 в NK-клетках, кажется, что нет какой-либо особой субпопуляции. . . . .
VlnPlot(NK_subtype,features = c('KLRF1','IL7R','ZNF683','TPSAB1',
'LGALS9','CD44','PTPRC','HAVCR2','NKG7','TIGIT',
'LAG3','CTLA4'),group.by = 'type',pt.size = 0)
Анализ пролиферации лимфы при GSE150825 рините
Аналогично, давайте посмотрим на NK-клетки в наборе данных GSE150825 по лимфоидной гиперплазии при рините.
# 150825
data_150825 <- CreateSeuratObject(counts = Read10X('GSE150825_RAW/rawdata/'),
min.cells = 3, min.features = 200)
dim(data_150825)
head(data_150825@meta.data)
# Уменьшение размерности
data_150825 <- NormalizeData(data_150825)
data_150825 <- FindVariableFeatures(data_150825)
data_150825 <- ScaleData(data_150825)
data_150825 <- RunPCA(data_150825)
ElbowPlot(data_150825,ndims = 30)
# Группировка
data_150825 <- RunTSNE(data_150825,reduction = 'pca',dims = 1:10)
DimPlot(data_150825,reduction = 'tsne',label = T)
data_150825 <- FindNeighbors(data_150825,dims = 1:10)
data_150825 <- FindClusters(data_150825,resolution = 0.6)
DimPlot(data_150825,reduction = 'tsne',label = T,group.by = 'seurat_clusters')
DotPlot(data_150825,features = c('CD14', 'CD68', 'CD163', 'ITGAX','FCGR3A'))
cell_type <- data.frame(cluster = 0:19,type = 0:19)
cell_type[cell_type$cluster %in% c(1,3,4,5,7,9),'type'] <- 'T cells'
cell_type[cell_type$cluster %in% c(3,5,7,11),'type'] <- 'NK cell'
cell_type[cell_type$cluster %in% c(8,18),'type'] <- 'Myeloid cells'
cell_type[cell_type$cluster %in% c(13,17),'type'] <- 'Epithelial cells'
cell_type[cell_type$cluster %in% c(0,2,6,10,12,16),'type'] <- 'B cells'
cell_type[cell_type$cluster %in% c(6,19),'type'] <- 'plasma cells'
cell_type[cell_type$cluster %in% c(14),'type'] <- 'mast cells'
cell_type[cell_type$cluster %in% c(15),'type'] <- 'fiberblast'
data_150825@meta.data$type <- unlist(lapply(data_150825@meta.data$seurat_clusters,function(x){cell_type[cell_type$cluster==x,'type']}))
DimPlot(data_150825,reduction = 'tsne',group.by = 'type',label = T)
data_150825_nk <- subset(data_150825,subset= type == 'NK cell')
data_150825_nk <- NormalizeData(data_150825_nk)
data_150825_nk <- FindVariableFeatures(data_150825_nk)
data_150825_nk <- ScaleData(data_150825_nk)
data_150825_nk <- RunPCA(data_150825_nk)
ElbowPlot(data_150825_nk,ndims = 30)
data_150825_nk <- RunTSNE(data_150825_nk,reduction = 'pca',dims = 1:20)
DimPlot(data_150825_nk,reduction = 'tsne')
data_150825_nk <- FindNeighbors(data_150825_nk,dims = 1:20)
data_150825_nk <- FindClusters(data_150825_nk,resolution = 0.1)
DimPlot(data_150825_nk,reduction = 'tsne',group.by = 'seurat_clusters',label = T)
data_150825_nk$type <- sapply(data_150825_nk$seurat_clusters, function(x){paste0('NK',as.numeric(x))})
DimPlot(data_150825_nk,reduction = 'tsne',group.by = 'type',label = T)
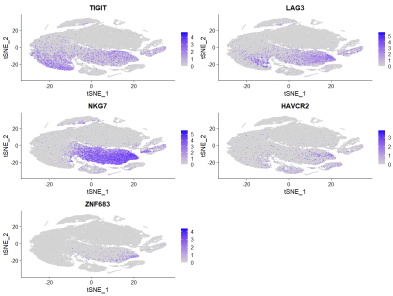
FeaturePlot(data_150825,features = c('TIGIT','LAG3','NKG7','HAVCR2','ZNF683'))
Очень странноВ этом наборе данных не было найдено соответствующей информации о группировке, а также группа NLH и группа NPC не были найдены.
Следующий проходГлобальная клеточная связь, чтобы увидеть, какие клетки взаимодействуют с NK-клетками
# all cellchat
data.input <- data_162025@assays$RNA@data
meta.data <- data_162025@meta.data
cellchat <- createCellChat(object=data.input,
meta = meta.data,
group.by='type')
cellchat <- addMeta(cellchat,meta = meta.data)
cellchatDB <- CellChatDB.human
cellchat@DB <- cellchatDB
cellchat <- subsetData(cellchat)
cellchat <- identifyOverExpressedGenes(cellchat)
cellchat <- identifyOverExpressedInteractions(cellchat)
cellchat <- computeCommunProb(cellchat,population.size = F)
cellchat <- filterCommunication(cellchat,min.cells = 10)
df.net <- subsetCommunication(cellchat)
df.pathway <- subsetCommunication(cellchat,slot.name = 'netP')
cellchat <- computeCommunProbPathway(cellchat)
cellchat <- aggregateNet(cellchat)
netVisual_bubble(cellchat, sources.use = c(1,2,4,6),
targets.use = c(4,5,6),signaling = 'GALECTIN',remove.isolate = FALSE)
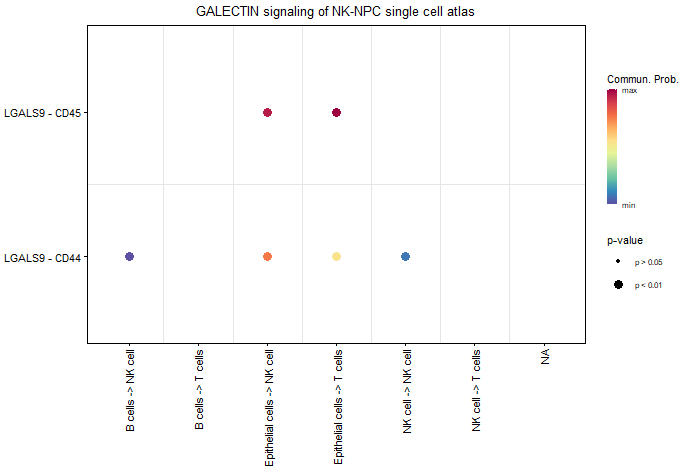
можно увидетьСигнальная связь между LGALS9-CD44 и LGALS9-CD45 между эпителиальными клетками и NK-клетками,и ранее не сообщалось,Итак, для эпителияклеткаруководитьсубпопуляция Сегментация
# Epithelial subtype
Epi_sub <- subset(data_162025,subset=type=='Epithelial cells')
Epi_sub <- NormalizeData(Epi_sub)
Epi_sub <- FindVariableFeatures(Epi_sub)
Epi_sub <- ScaleData(Epi_sub)
Epi_sub <- RunPCA(Epi_sub)
ElbowPlot(Epi_sub,ndims = 30)
Epi_sub <- RunTSNE(Epi_sub,reduction = 'pca',dims = 1:20)
DimPlot(Epi_sub,reduction = 'tsne')
Epi_sub <- FindNeighbors(Epi_sub,dims = 1:20)
Epi_sub <- FindClusters(Epi_sub,resolution = 0.3)
DimPlot(Epi_sub,reduction = 'tsne',group.by = 'seurat_clusters',label = T)
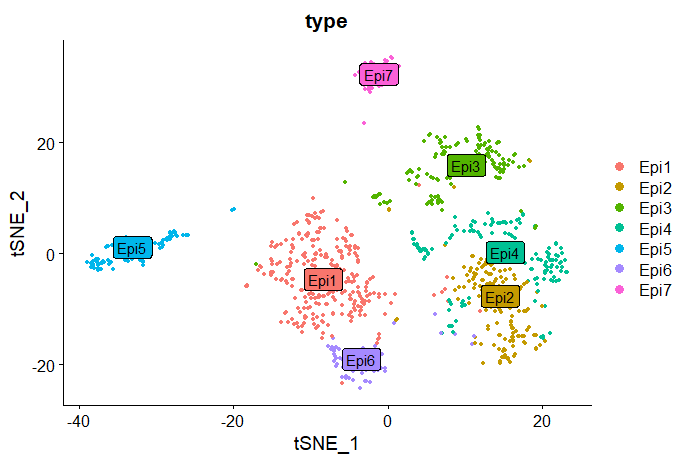
Epi_sub$type <- sapply(Epi_sub$seurat_clusters, function(x){paste0('Epi',as.numeric(x))})
DimPlot(Epi_sub,reduction = 'tsne',group.by = 'type',label = T,label.box = T)
dim(Epi_sub)
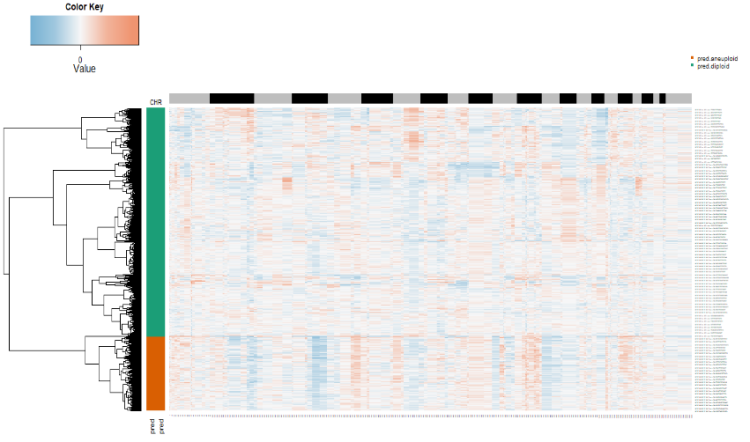
# copycat
# Определить нормальные клетки и опухолевые клетки
Epi_cnv <- copykat(rawmat=Epi_sub@assays$RNA@counts, id.type="S", ngene.chr=5, win.size=25,
KS.cut=0.1, sam.name="Epi_sub", distance="euclidean", norm.cell.names="",
output.seg="FLASE", plot.genes="TRUE", genome="hg20",n.cores=1)
saveRDS(Epi_cnv,file = 'Epi_cnv.RDS')
Epi_cnv <- readRDS('Epi_cnv.RDS')
pred.test <- data.frame(Epi_cnv$prediction)
pred.test <- pred.test[-which(pred.test$copykat.pred=="not.defined"),] ##remove undefined cells
CNA.test <- data.frame(Epi_cnv$CNAmat)
head(pred.test)
head(CNA.test[ , 1:5])
Epi_sub@meta.data$copykat.pred <- ifelse(rownames(Epi_sub@meta.data) %in% rownames(pred.test[pred.test$copykat.pred=='aneuploid',]),'aneuploid',
ifelse(rownames(Epi_sub@meta.data) %in% rownames(pred.test[pred.test$copykat.pred=='diploid',]),'diploid',NA)
)
my_palette <- colorRampPalette(rev(RColorBrewer::brewer.pal(n = 3, name = "RdBu")))(n = 999)
chr <- as.numeric(CNA.test$chrom) %% 2+1
rbPal1 <- colorRampPalette(c('black','grey'))
CHR <- rbPal1(2)[as.numeric(chr)]
chr1 <- cbind(CHR,CHR)
rbPal5 <- colorRampPalette(RColorBrewer::brewer.pal(n = 8, name = "Dark2")[2:1])
com.preN <- pred.test$copykat.pred
pred <- rbPal5(2)[as.numeric(factor(com.preN))]
cells <- rbind(pred,pred)
col_breaks = c(seq(-1,-0.4,length=50),seq(-0.4,-0.2,length=150),
seq(-0.2,0.2,length=600),seq(0.2,0.4,length=150),seq(0.4, 1,length=50))
heatmap.3(t(CNA.test[,4:ncol(CNA.test)]),dendrogram="r",
distfun = function(x) parallelDist::parDist(x,threads =4, method = "euclidean"),
hclustfun = function(x) hclust(x, method="ward.D2"),
ColSideColors=chr1,RowSideColors=cells,Colv=NA, Rowv=TRUE,
notecol="black",col=my_palette,breaks=col_breaks, key=TRUE,
keysize=1, density.info="none", trace="none",
cexRow=0.1,cexCol=0.1,cex.main=1,cex.lab=0.1,
symm=F,symkey=F,symbreaks=T,cex=1, cex.main=4, margins=c(10,10))
legend("topright", paste("pred.",names(table(com.preN)),sep=""), pch=15,col=RColorBrewer::brewer.pal(n = 8, name = "Dark2")[2:1], cex=0.6, bty="n")
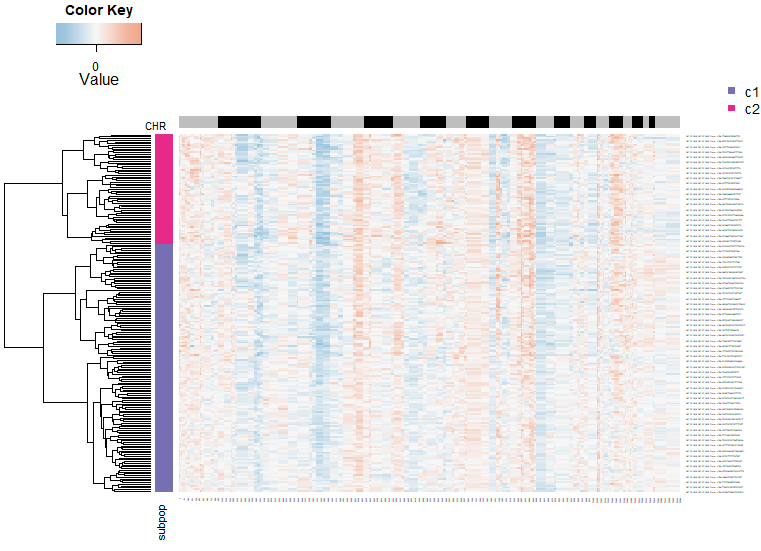
# Определить подтипы опухолевых клеток
tumor.cells <- pred.test$cell.names[which(pred.test$copykat.pred=="aneuploid")]
tumor.mat <- CNA.test[, which(colnames(CNA.test) %in% tumor.cells)]
hcc <- hclust(parallelDist::parDist(t(tumor.mat),threads =4, method = "euclidean"), method = "ward.D2")
hc.umap <- cutree(hcc,2)
rbPal6 <- colorRampPalette(RColorBrewer::brewer.pal(n = 8, name = "Dark2")[3:4])
subpop <- rbPal6(2)[as.numeric(factor(hc.umap))]
cells <- rbind(subpop,subpop)
heatmap.3(t(tumor.mat),dendrogram="r", distfun = function(x) parallelDist::parDist(x,threads =4, method = "euclidean"), hclustfun = function(x) hclust(x, method="ward.D2"),
ColSideColors=chr1,RowSideColors=cells,Colv=NA, Rowv=TRUE,
notecol="black",col=my_palette,breaks=col_breaks, key=TRUE,
keysize=1, density.info="none", trace="none",
cexRow=0.1,cexCol=0.1,cex.main=1,cex.lab=0.1,
symm=F,symkey=F,symbreaks=T,cex=1, cex.main=4, margins=c(10,10))
legend("topright", c("c1","c2"), pch=15,col=RColorBrewer::brewer.pal(n = 8, name = "Dark2")[3:4], cex=0.9, bty='n')
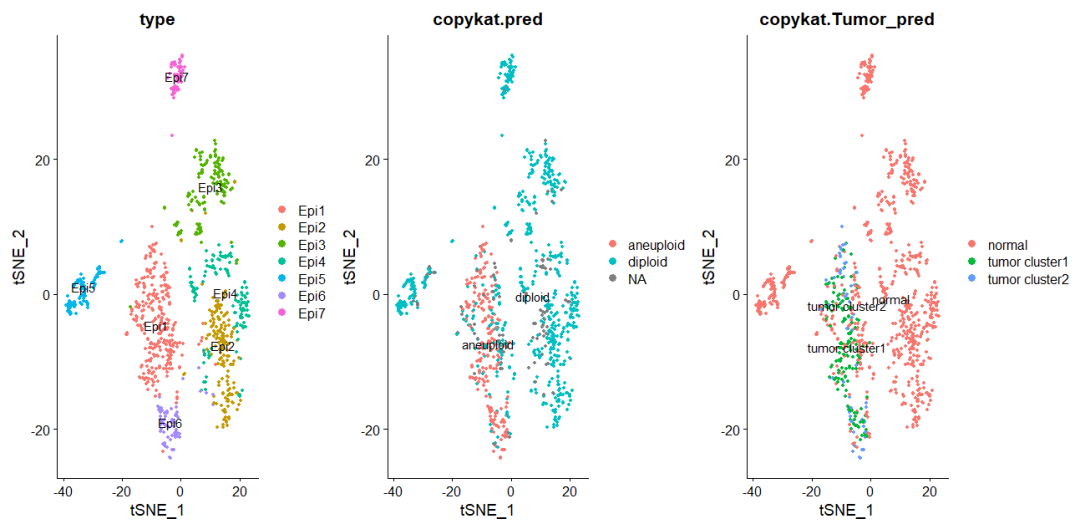
Epi_sub@meta.data$copykat.Tumor_pred <- ifelse(colnames(Epi_sub) %in% names(hc.umap[hc.umap==1]),'tumor cluster1',
ifelse(colnames(Epi_sub) %in% names(hc.umap[hc.umap==2]),'tumor cluster2','normal'))
p1 <- DimPlot(Epi_sub,group.by = 'type',label = T)
p2 <- DimPlot(Epi_sub,group.by = 'copykat.pred',label = T)
p3 <- DimPlot(Epi_sub,group.by = 'copykat.Tumor_pred',label=T)
p1+p2+p3
По результатам идентификации копикатов можно различить нормальные клетки и опухоли, но два подтипа опухолевых клеток кажутся смешанными.
Визуализация маркерных генов
DotPlot(Epi_sub,features = c('LGALS9','HLA-A','HLA-B','HLA-C','HLA-E','HLA-F','HLA-G','B2M'),group.by = 'copykat.Tumor_pred')
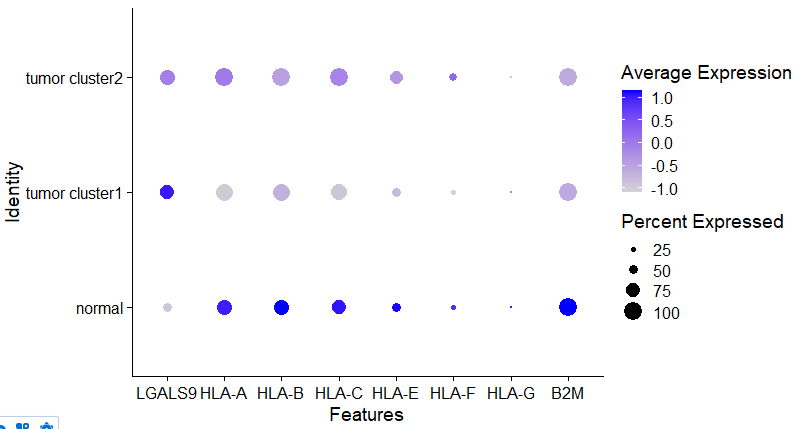
NK-клетки могут распознавать и уничтожать опухолевые клетки посредством подавления MHC класса I. Можно видеть, что в обоих типах опухолевых клеток молекулы MHC класса I подавлены.
Здесь экспрессия LGALS9 в кластере 1 выше, чем в кластере 2, что несколько похоже на подтип 2 в статье и может вызывать истощение NK-клеток.
Давайте посмотрим на экспрессию белков, связанных с инфекцией EBV, в двух типах клеток.
FeaturePlot(Epi_sub,features = c('KRT5','SSTR2','LMP-1/BNLF2a/b','RPMS1/A73'))
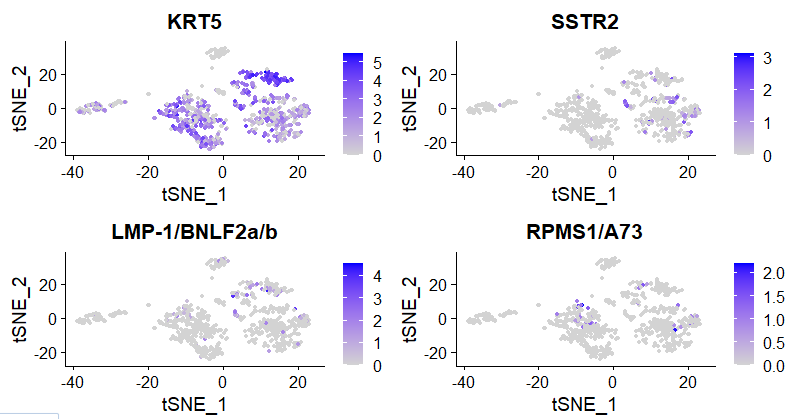
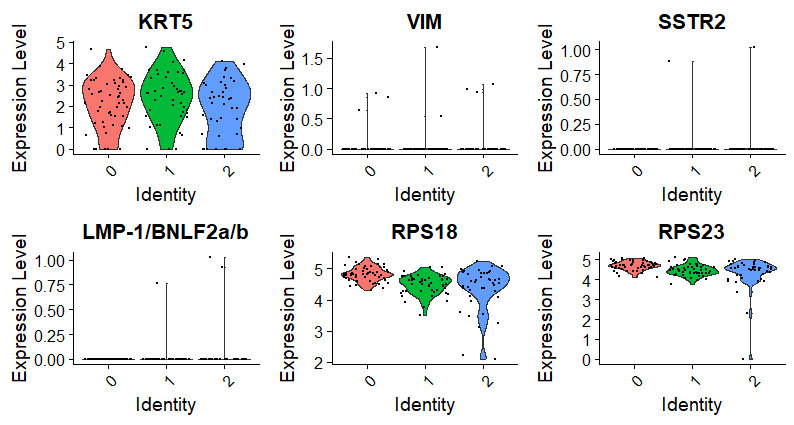
Опухоль1 и опухоль2 здесь не так разделены, как в оригинальном тексте, поэтому судить о разнице между двумя типами опухолевых клеток невозможно.
Далее автор посчитал, что статус активности ВЭБ может влиять на опухоль1, поэтому он выделил опухоль1 для квазихронологического анализа. Здесь я просто случайным образом выбрал подгруппу.
# tumor1 субпопуляция
tumor1_sub <- subset(tumor1_sub,subset=copykat.Tumor_pred=='tumor cluster1')
tumor1_sub <- NormalizeData(tumor1_sub)
tumor1_sub <- FindVariableFeatures(tumor1_sub)
tumor1_sub <- ScaleData(tumor1_sub)
tumor1_sub <- RunPCA(tumor1_sub)
ElbowPlot(tumor1_sub,ndims = 30)
tumor1_sub <- RunTSNE(tumor1_sub,reduction = 'pca',dims = 1:20)
DimPlot(tumor1_sub,reduction = 'tsne')
tumor1_sub <- FindNeighbors(tumor1_sub,dims = 1:20)
tumor1_sub <- FindClusters(tumor1_sub,resolution = 0.8)
DimPlot(tumor1_sub,reduction = 'tsne',group.by = 'seurat_clusters',label = T)
tumor1_sub$type <- sapply(tumor1_sub$seurat_clusters, function(x){paste0('Epi',as.numeric(x))})
# SCORPIUS Квази-тайминг
group_name <- as.numeric(as.factor(tumor1_sub@meta.data$type))
names(group_name) <- tumor1_sub@meta.data$type
group_name <- as.factor(group_name)
space <- reduce_dimensionality(t(tumor1_sub@assays$RNA@data), "spearman")
draw_trajectory_plot(space,group_name, contour = TRUE)
traj <- infer_trajectory(space)
draw_trajectory_plot(space, group_name, traj$path, contour = TRUE)
gimp <- gene_importances(
t(tumor1_sub@assays$RNA@scale.data),
traj$time,
num_permutations = 10,
num_threads = 8,
ntree = 10000,
ntree_perm = 1000
)
gimp$qvalue <- p.adjust(gimp$pvalue, "BH", length(gimp$pvalue))
gene_sel <- gimp$gene[gimp$qvalue < .05]
expr_sel <- scale_quantile(t(tumor1_sub@assays$RNA@scale.data)[,gene_sel])
time <- traj$time
draw_trajectory_heatmap(expr_sel, time,progression_group=group_name)
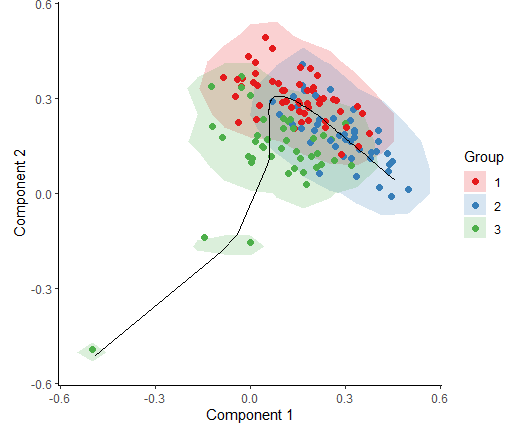
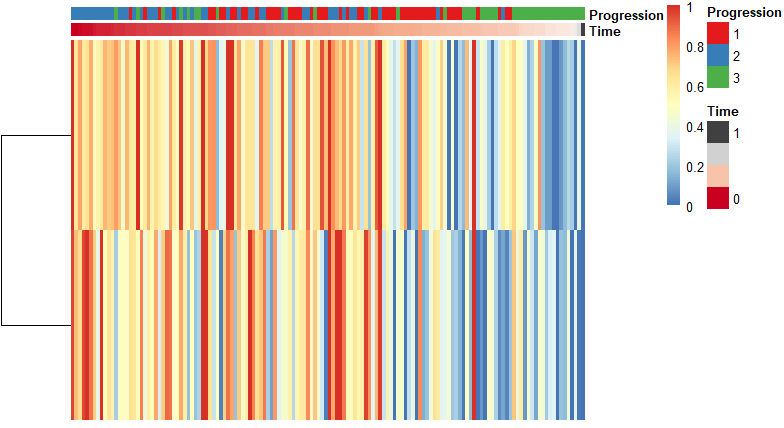
Как видите, эффект от дальнейшего разделения не очень хороший. Давайте проследим за процессом.
# tumor1 разница
tumor1_sub_markers <- FindAllMarkers(tumor1_sub)
tumor1_sub_markers$cluster <- sapply(tumor1_sub_markers$cluster, function(x){paste0('Epi',as.numeric(x))})
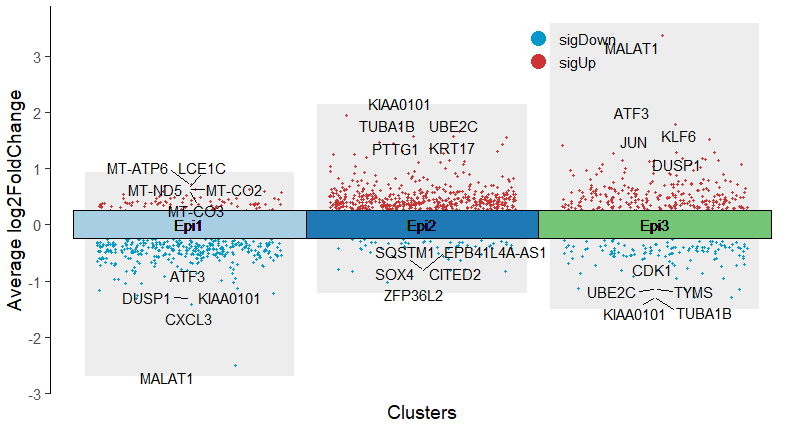
jjVolcano(tumor1_sub_markers)
VlnPlot(tumor1_sub,features = c('KRT5','VIM','SSTR2','LMP-1/BNLF2a/b','RPS18','RPS23'))
tumor1_sub_deg <- tumor1_sub_markers[tumor1_sub_markers$cluster=='Epi1',]
tumor1_sub_marker <- tumor1_sub_deg$avg_log2FC
names(tumor1_sub_marker) <- bitr(tumor1_sub_deg$gene,fromType = 'SYMBOL',toType = 'ENTREZID',OrgDb = org.Hs.eg.db)[,'ENTREZID']
tumor1_sub_marker <- sort(tumor1_sub_marker,decreasing = T)
tumor1 <- gseKEGG(tumor1_sub_marker)
gseaplot2(tumor1,geneSetID = 2)



Углубленный анализ переполнения памяти CUDA: OutOfMemoryError: CUDA не хватает памяти. Попыталась выделить 3,21 Ги Б (GPU 0; всего 8,00 Ги Б).
[Решено] ошибка установки conda. Среда решения: не удалось выполнить первоначальное зависание. Повторная попытка с помощью файла (графическое руководство).
Прочитайте нейросетевую модель Трансформера в одной статье

.ART Теплые зимние предложения уже открыты

Сравнительная таблица описания кодов ошибок Amap

Уведомление о последних правилах Points Mall в декабре 2022 года.

Даже новички могут быстро приступить к работе с легким сервером приложений.
Взгляд на RSAC 2024|Защита конфиденциальности в эпоху больших моделей
Вы используете ИИ каждый день и до сих пор не знаете, как ИИ дает обратную связь? Одна статья для понимания реализации в коде Python общих функций потерь генеративных моделей + анализ принципов расчета.

Используйте (внутренний) почтовый ящик для образовательных учреждений, чтобы использовать Microsoft Family Bucket (1T дискового пространства на одном диске и версию Office 365 для образовательных учреждений)
Руководство по началу работы с оперативным проектом (7) Практическое сочетание оперативного письма — оперативного письма на основе интеллектуальной системы вопросов и ответов службы поддержки клиентов

[docker] Версия сервера «Чтение 3» — создайте свою собственную программу чтения веб-текста

Обзор Cloud-init и этапы создания в рамках PVE
Корпоративные пользователи используют пакет регистрационных ресурсов для регистрации ICP для веб-сайта и активации оплаты WeChat H5 (с кодом платежного узла версии API V3)

Подробное объяснение таких показателей производительности с высоким уровнем параллелизма, как QPS, TPS, RT и пропускная способность.
Удачи в конкурсе Python Essay Challenge, станьте первым, кто испытает новую функцию сообщества [Запускать блоки кода онлайн] и выиграйте множество изысканных подарков!

[Техническая посадка травы] Кровавая рвота и отделка позволяют вам необычным образом ощипывать гусиные перья! Не распространяйте информацию! ! !

[Официальное ограниченное по времени мероприятие] Сейчас ноябрь, напишите и получите приз

Прочтите это в одной статье: Учебник для няни по созданию сервера Huanshou Parlu на базе CVM-сервера.

Cloud Native | Что такое CRD (настраиваемые определения ресурсов) в K8s?

Как использовать Cloudflare CDN для настройки узла (CF самостоятельно выбирает IP) Гонконг, Китай/Азия узел/сводка и рекомендации внутреннего высокоскоростного IP-сегмента

Дополнительные правила вознаграждения амбассадоров акции в марте 2023 г.
Можно ли открыть частный сервер Phantom Beast Palu одним щелчком мыши? Супер простой урок для начинающих! (Прилагается метод обновления сервера)
[Играйте с Phantom Beast Palu] Обновите игровой сервер Phantom Beast Pallu одним щелчком мыши

Maotouhu делится: последний доступный внутри страны адрес склада исходного образа Docker 2024 года (обновлено 1 декабря)

Кодирование Base64 в MultipartFile
5 точек расширения SpringBoot, супер практично!

Глубокое понимание сопоставления индексов Elasticsearch.
15 рекомендуемых платформ разработки с нулевым кодом корпоративного уровня. Всегда найдется та, которая вам понравится.
