Прямая трансляция seurat v5, выполните интеграцию пяти данных одним щелчком мыши: Harmony, CCA, RPCA, FastMNN, scVI
Удачи в году Дракона! Я очень рад встретиться со всеми вами в первый день первого лунного месяца.
В этой статье в основном тестируются: пять методов интеграции отдельных ячеек в среде seuratv5.
- · Интеграция метода CCA
- ·Интеграция метода RPCA
- · Интеграция метода Гармонии
- · Интеграция методов FastMNN
- · интеграция метода scVI

Доктор Шэнсинь
[Биоинформатика] Язык R, изучение биоинформатики, серат, секвенирование одиночных клеток, пространственный транскриптом. Python, scanpy, cell2location, обмен данными
- Сначала загрузите pbmc в среде v5.
2 #https://satijalab.org/seurat/articles/install_v5.html#2Install v5---.libPaths( в папке seurat_v5 c( '/home/rootyll/seurat_v5/', "/usr/local/lib/R/site-library", "/usr/lib/R/site-library", "/usr/lib/R/library" ))
library(Seurat)pbmc = readRDS('~/gzh/pbmc3k_final.rds')
DimPlot(pbmc)- Мы видим, что объект pbmc в настоящее время все еще является объектом seurat v4.

- Шаг 2. Преобразуйте объект seuratv4 в объект seuratv5, чтобы реализовать сосуществование объекта seuratv4 и объекта v5.:Используйте seuratv5 для чтения файлов rds, созданных seurat v4.
pbmc[["RNA5"]] <- as(object = pbmc[["RNA"]], Class = "Assay5")
DefaultAssay(pbmc)Assays(pbmc)
pbmc[["RNA_seuratv4"]] <- pbmc[['RNA']]
pbmc[['RNA']]=pbmc[['RNA5']]
- Если это один образец, просто следуйте приведенной ниже стандартной процедуре Сёра. Разницы между стандартными процессами v4 и v5 нет.
#3 Стандартный процесс объекта v5 ----#Добавьте столбец данных процентов.mt в pbmc
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
#Визуализируйте показатели контроля качества с помощью скрипичных графиков
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
#FeatureScatter часто используется для визуализации. feature-feature Актуальность, #nCount_RNA Соответствие проценту.mt
plot1 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "percent.mt")
Корреляция между #nCount_RNA и nFeature_RNA
plot2 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
plot1 + plot2 #Объединить два изображения
pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)
#выбирать 2500 > nFeature_RNA >200 и процент.мт < 5 данных
pbmc <- NormalizeData(object = pbmc)pbmc <- FindVariableFeatures(object = pbmc)
pbmc <- ScaleData(object = pbmc)
pbmc <- RunPCA(object = pbmc)
pbmc <- FindNeighbors(object = pbmc, dims = 1:30)
pbmc <- FindClusters(object = pbmc)
pbmc <- RunUMAP(object = pbmc, dims = 1:30)
DimPlot(object = pbmc, reduction = "umap")Результаты следующие:
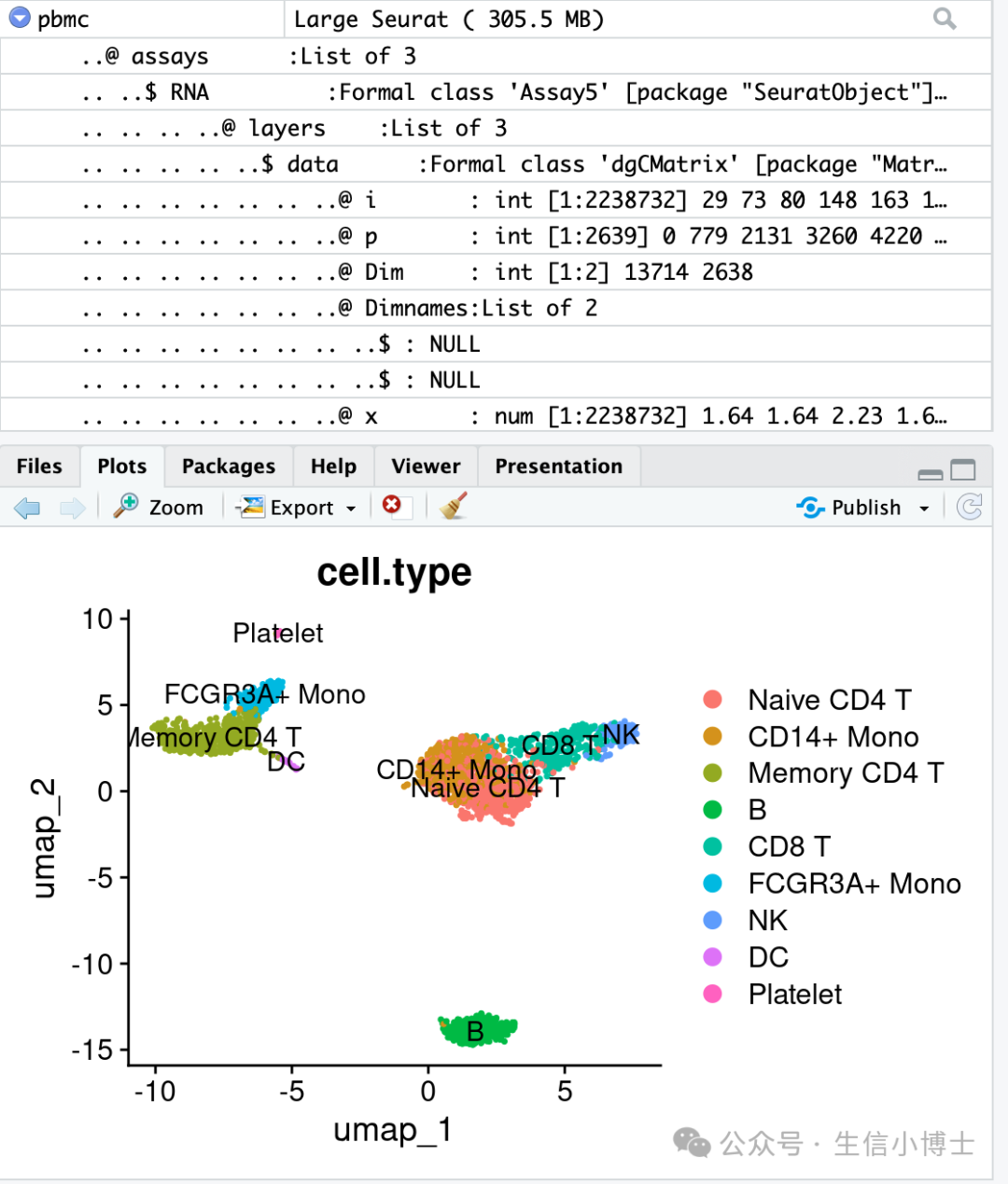
- Если обрабатывается несколько образцов, требуется интеграция образцов. Версия seuratv5 объединяет 5 методов интеграции. Давайте попробуем их:
- Во-первых, нам нужно создать два объекта seuratv5.
#для демонстрации,Делим объект pbmc на 2 набора данных,Выполните комплексный анализ
dim(pbmc)#13714 2638
pbmc$group= ifelse(pbmc$nCount_RNA>2200,yes = "CTRL",no = 'STIM')
table(pbmc$group)4##4 # In line with prior workflows, you can also into split your object into a list of multiple objects based on a metadata# column creates a list of two objects
ifnb_list <- SplitObject(pbmc, split.by = "group")
ifnb_list$CTRLifnb_list$STIM
- После получения двух объектов Seuart проводится комплексный анализ.
- · Интеграция метода CCA
- ·Интеграция метода RPCA
- · Интеграция метода Гармонии
- · Интеграция методов FastMNN
- · интеграция метода scVI
- Из-за нехватки времени здесь используются только методы интеграции cca, rpca и гармонии.
#4 У нас есть два объекта seuratv5 для комплексного анализа ------merged_obj <- merge(x = ifnb_list$CTRL, y = ifnb_list$STIM)merged_obj <- NormalizeData(merged_obj)merged_obj <- FindVariableFeatures(merged_obj)merged_obj <- ScaleData(merged_obj)merged_obj <- RunPCA(merged_obj)obj=merged_obj#rpcaobj <- IntegrateLayers(object = obj, method = RPCAIntegration, orig.reduction = "pca", new.reduction = "integrated.rpca", verbose = FALSE)#ccaobj <- IntegrateLayers( object = obj, method = CCAIntegration, orig.reduction = "pca", new.reduction = "integrated.cca", verbose = FALSE)#remotes::install_github("satijalab/seurat-wrappers")#BiocManager::install('batchelor')#BiocManager::install('SeuratData',force = TRUE)# obj <- IntegrateLayers(# object = obj, method = FastMNNIntegration,# new.reduction = "integrated.mnn",# verbose = FALSE# )# # SeuratWrappers::RunFastMNN(object.list = obj,reduction.name = 'mnn')#harmonyobj <- IntegrateLayers( object = obj, method = HarmonyIntegration, orig.reduction = "pca", new.reduction = "harmony", verbose = FALSE)- Здесь показаны только результаты интеграции cca и гармонии.
obj <- FindNeighbors(obj, reduction = "integrated.cca", dims = 1:30)
obj <- FindClusters(obj, resolution = 2, cluster.name = "cca_clusters")
obj <- RunUMAP(obj, reduction = "integrated.cca", dims = 1:30, reduction.name = "umap.cca")
p1 <- DimPlot( obj, reduction = "umap.cca", group.by = c("Method", "predicted.celltype.l2", "cca_clusters"), combine = FALSE, label.size = 2)
obj <- FindNeighbors(obj, reduction = "harmony", dims = 1:30)obj <- FindClusters(obj, resolution = 2, cluster.name = "harmony_clusters")
obj <- RunUMAP(obj, reduction = "harmony", dims = 1:30, reduction.name = "harmony")
p2 <- DimPlot( obj, reduction = "harmony", group.by = c("Method", "cell.type", "harmony_clusters"), combine = FALSE, label.size = 2)
library(patchwork)
wrap_plots(c(p1, p2), ncol = 2, byrow = F)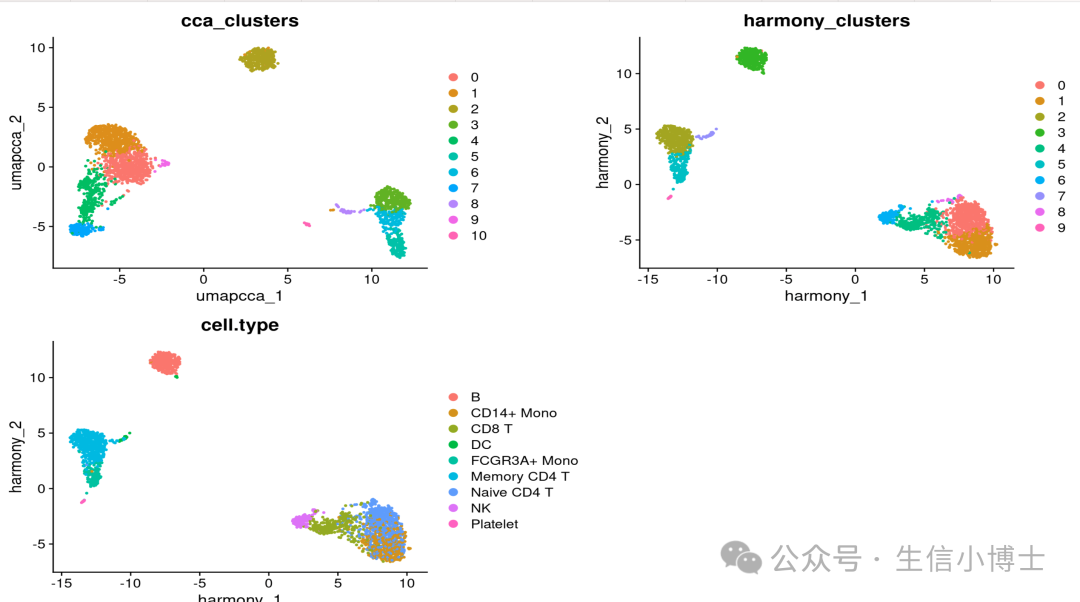
Кажется, с согласованностью все в порядке, но в будущем есть 5 способов транслировать этот твит в прямом эфире, чтобы увидеть, какой метод быстрее и лучше.
ссылка:
- Стандартный процесс seuratv5: https://satijalab.org/seurat/articles/essential_commands
- Разница между seuratv4 и seruatv5: https://satijalab.org/seurat/articles/announcements.html
- Анализ интеграции Seratv5: https://satijalab.org/seurat/articles/seurat5_integration https://satijalab.org/seurat/articles/integration_introduction



Углубленный анализ переполнения памяти CUDA: OutOfMemoryError: CUDA не хватает памяти. Попыталась выделить 3,21 Ги Б (GPU 0; всего 8,00 Ги Б).
[Решено] ошибка установки conda. Среда решения: не удалось выполнить первоначальное зависание. Повторная попытка с помощью файла (графическое руководство).
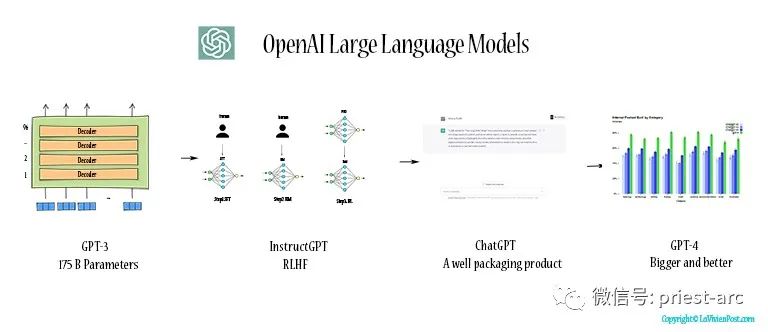
Прочитайте нейросетевую модель Трансформера в одной статье

.ART Теплые зимние предложения уже открыты

Сравнительная таблица описания кодов ошибок Amap

Уведомление о последних правилах Points Mall в декабре 2022 года.

Даже новички могут быстро приступить к работе с легким сервером приложений.
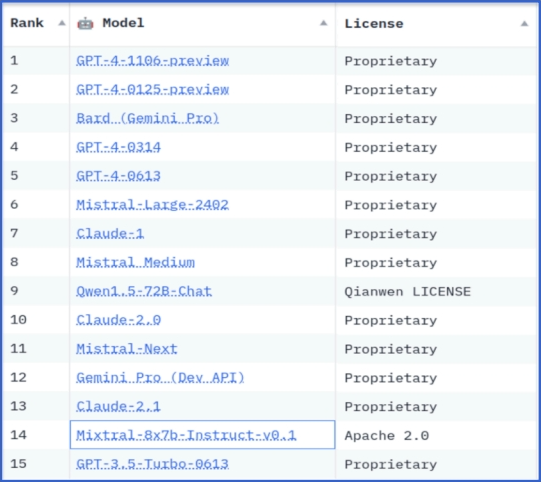
Взгляд на RSAC 2024|Защита конфиденциальности в эпоху больших моделей
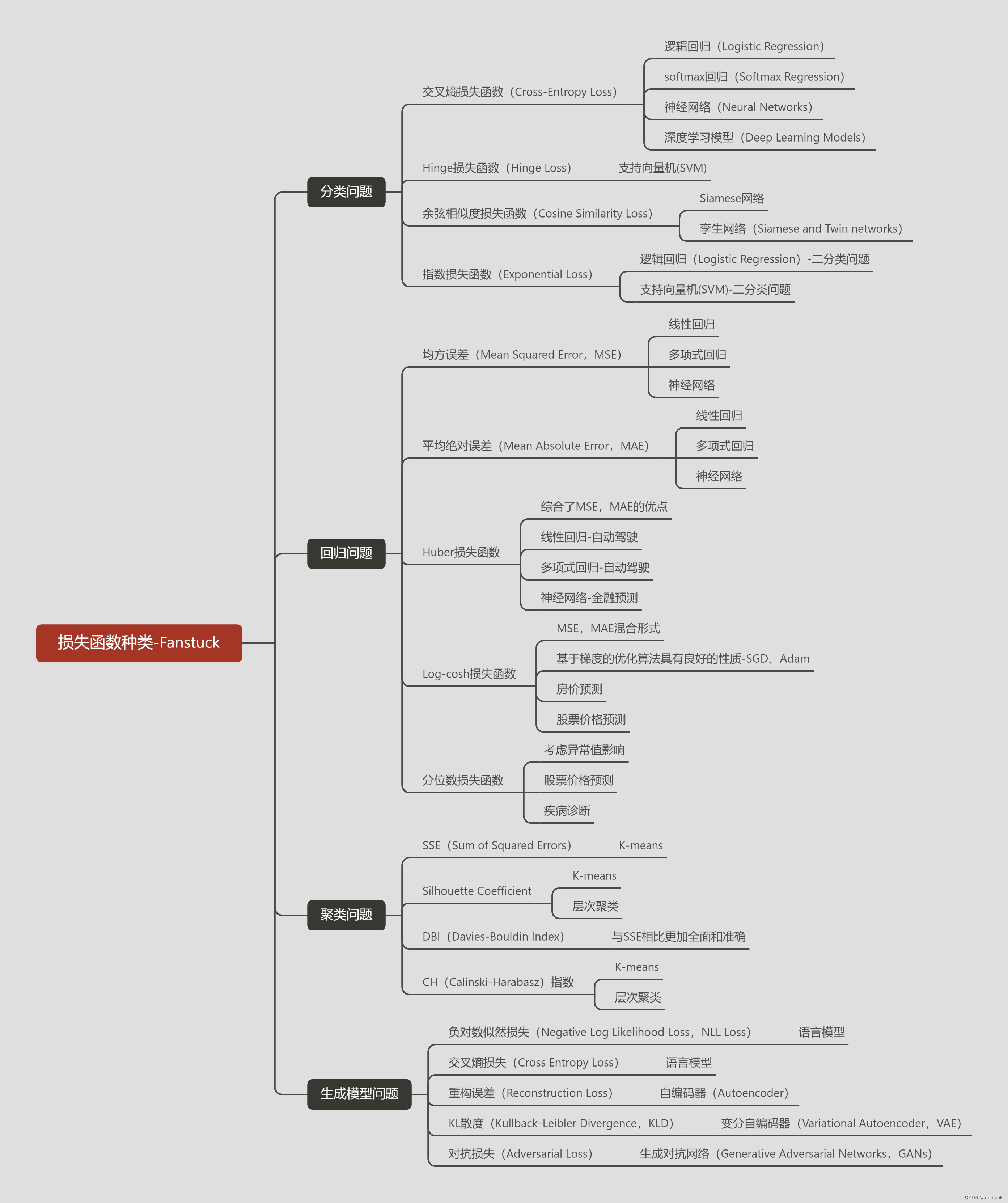
Вы используете ИИ каждый день и до сих пор не знаете, как ИИ дает обратную связь? Одна статья для понимания реализации в коде Python общих функций потерь генеративных моделей + анализ принципов расчета.

Используйте (внутренний) почтовый ящик для образовательных учреждений, чтобы использовать Microsoft Family Bucket (1T дискового пространства на одном диске и версию Office 365 для образовательных учреждений)
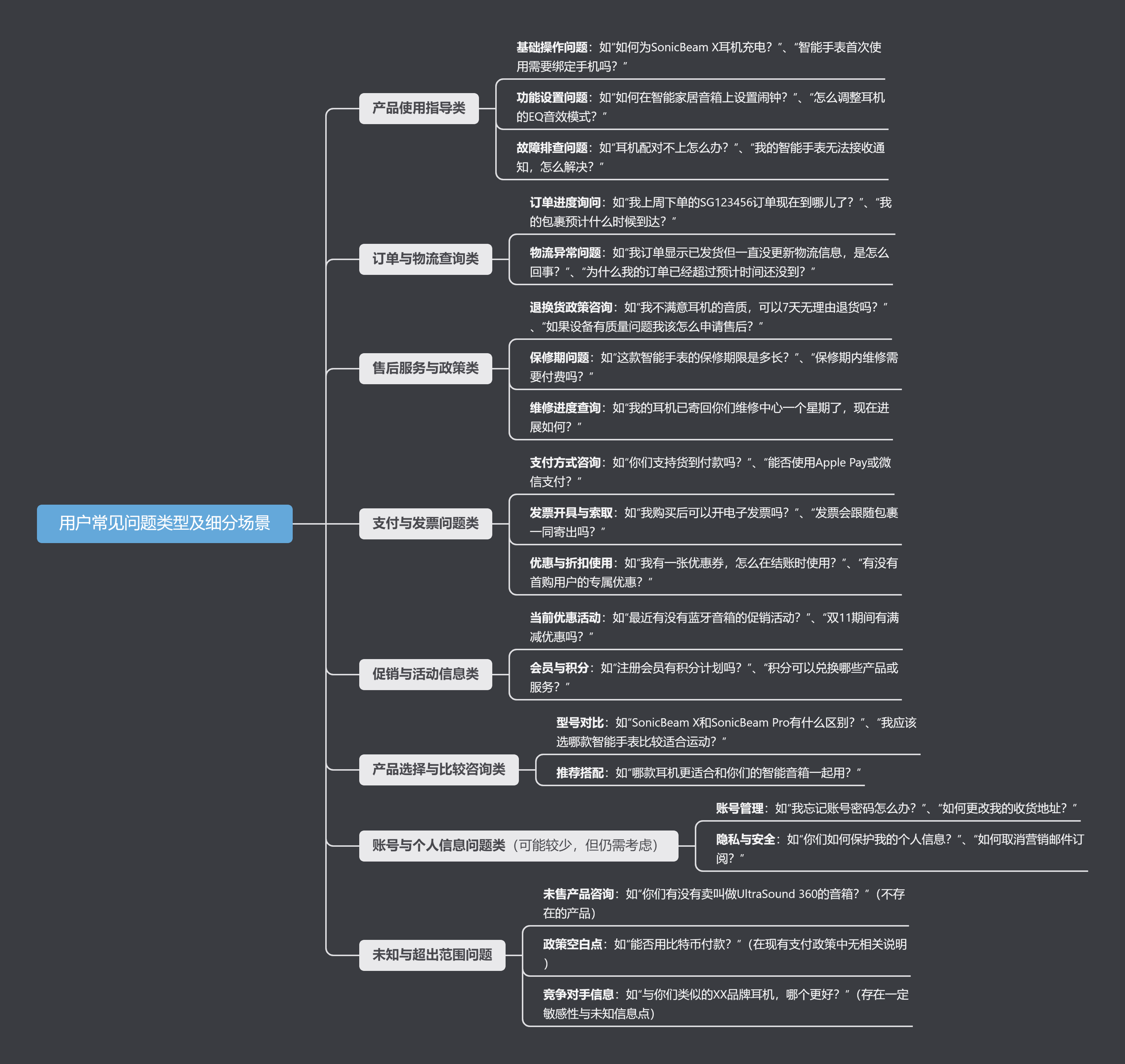
Руководство по началу работы с оперативным проектом (7) Практическое сочетание оперативного письма — оперативного письма на основе интеллектуальной системы вопросов и ответов службы поддержки клиентов

[docker] Версия сервера «Чтение 3» — создайте свою собственную программу чтения веб-текста

Обзор Cloud-init и этапы создания в рамках PVE
Корпоративные пользователи используют пакет регистрационных ресурсов для регистрации ICP для веб-сайта и активации оплаты WeChat H5 (с кодом платежного узла версии API V3)

Подробное объяснение таких показателей производительности с высоким уровнем параллелизма, как QPS, TPS, RT и пропускная способность.
Удачи в конкурсе Python Essay Challenge, станьте первым, кто испытает новую функцию сообщества [Запускать блоки кода онлайн] и выиграйте множество изысканных подарков!

[Техническая посадка травы] Кровавая рвота и отделка позволяют вам необычным образом ощипывать гусиные перья! Не распространяйте информацию! ! !

[Официальное ограниченное по времени мероприятие] Сейчас ноябрь, напишите и получите приз

Прочтите это в одной статье: Учебник для няни по созданию сервера Huanshou Parlu на базе CVM-сервера.

Cloud Native | Что такое CRD (настраиваемые определения ресурсов) в K8s?

Как использовать Cloudflare CDN для настройки узла (CF самостоятельно выбирает IP) Гонконг, Китай/Азия узел/сводка и рекомендации внутреннего высокоскоростного IP-сегмента

Дополнительные правила вознаграждения амбассадоров акции в марте 2023 г.
Можно ли открыть частный сервер Phantom Beast Palu одним щелчком мыши? Супер простой урок для начинающих! (Прилагается метод обновления сервера)
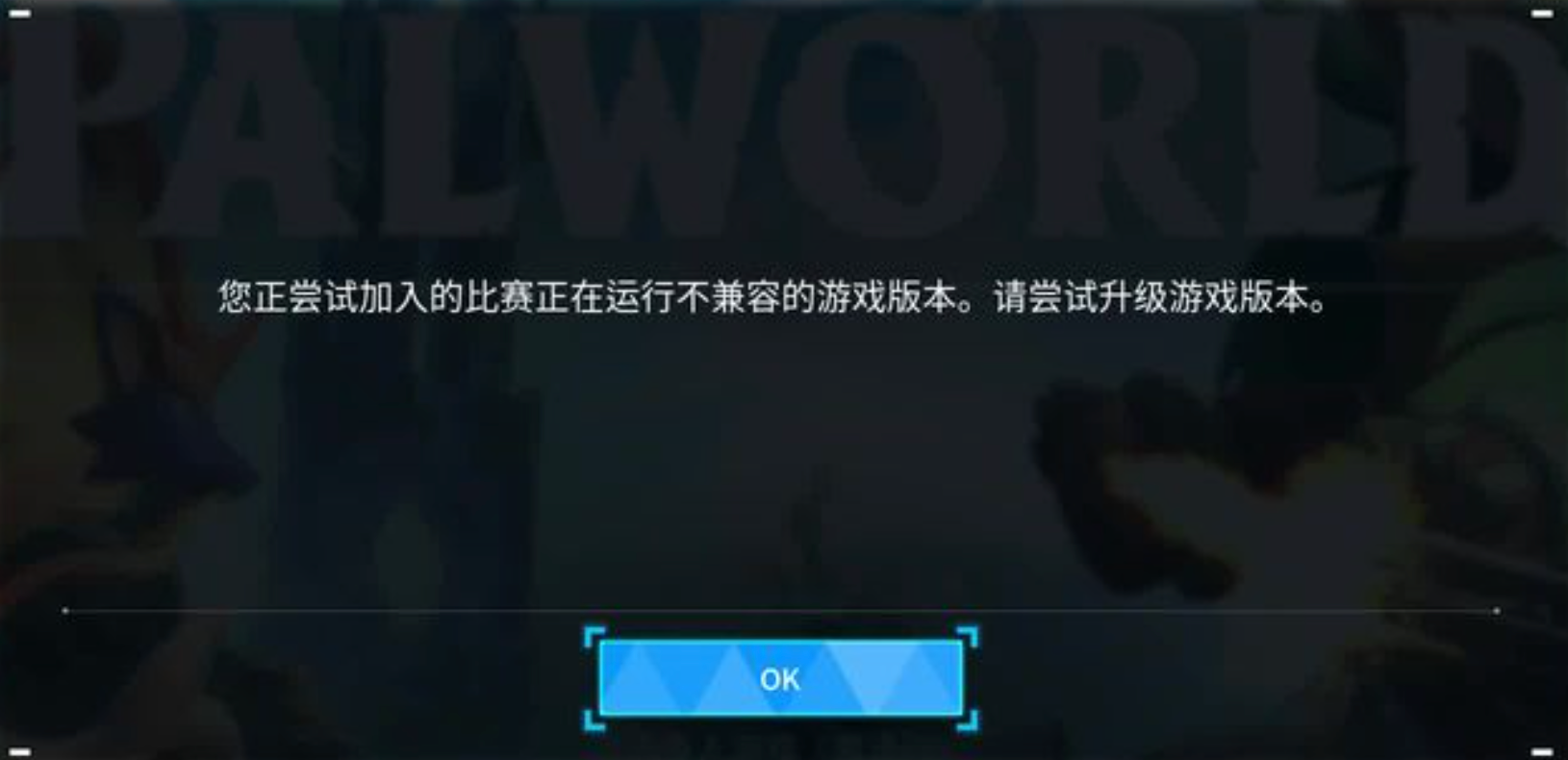
[Играйте с Phantom Beast Palu] Обновите игровой сервер Phantom Beast Pallu одним щелчком мыши

Maotouhu делится: последний доступный внутри страны адрес склада исходного образа Docker 2024 года (обновлено 1 декабря)

Кодирование Base64 в MultipartFile
5 точек расширения SpringBoot, супер практично!

Глубокое понимание сопоставления индексов Elasticsearch.
15 рекомендуемых платформ разработки с нулевым кодом корпоративного уровня. Всегда найдется та, которая вам понравится.
