Практическое исследование влияния пяти параметров на визуализацию графа UMAP
Эта статья《A comprehensive single-cell map of T cell exhaustion-associated immune environ- ments in human breast cancer》изUMAPНа картинкеTклеткаиBклеткаотделениз,Но когда мы снова появились раньше, мы обнаружили, что Т-клетки и В-клетки были вместе. Многие друзья недоумевают, почему фотографии и оригиналы документов, которые я воспроизвел, так отличаются. В общем, не стоит слишком нервничать,Есть параметры, которые влияют только невооруженным глазом, но не влияют на существенные свойства ячеек (то есть группировку). Но если вы хотите быть более серьезным, вы все равно можете изучить влияние параметров.
Цель этого твита — изучить влияние некоторых важных параметров на последующую визуализацию кластеризации UMAP. Основными параметрами, которые следует учитывать, являются: количество гипервариабельных генов; размерность pca; параметры n_neighbours, min_dist и dims в UMAP. Воздействие главным образом зависит от того, разделены ли Т-клетки и В-клетки.
Базовые знания о маркерных генах:
- Т-клетки (CD3D, CD3E)
- В-клетки (MS4A1, CD79A)
Определение параметра
- Параметр nfeatures FindVariableFeatures (количество гипервариабельных генов)
- параметр npcs RunPCA (размер PCA)
- RunUMAP из n.neighbors и min.dist параметр
- n_neighbors -- Используется для построения исходного многомерного графа на основе приблизительного числа точек ближайшего соседа. Он эффективно контролирует, как UMAP балансирует локальную и глобальную структуру — низкое значение заставит UMAP учитывать количество соседей с помощью ограничений при анализе многомерных данных, что требует большего внимания. наместная структура,Высокие значения заставят UMAP представлять общую структуру.,При этом теряются мелкие детали.
- min_dist — минимальное расстояние между точками в низкомерном пространстве. Этот параметр определяет, насколько близко друг к другу расположены кластеры UMAP.,Чем ниже значение,Точки встраивания более жесткие. Большие значения изmin_dist заставят UMAP более свободно упаковывать точки вместе.,Лучше сосредоточьтесь на сохранении широкого спектра топологий.
- dims -- Рассмотрим размер объекта (который dimensions to use as input features, used only if features is NULL)
Давайте посмотрим на влияние изменения переменной параметра:
Влияние гипервариабельных генов
Число гипервариабельных генов составляет 3000:
Конкретные параметры:
- 3000 гипервариабельных генов;
- размер ПКА 110;
- Параметры UMAP: n_neighbours — 50, min_dist — 0,1, dims — 1:15.
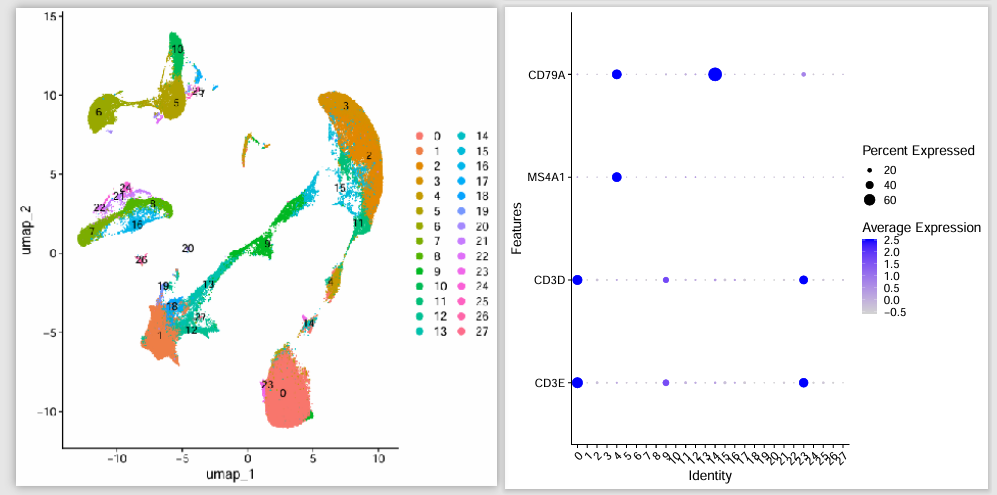
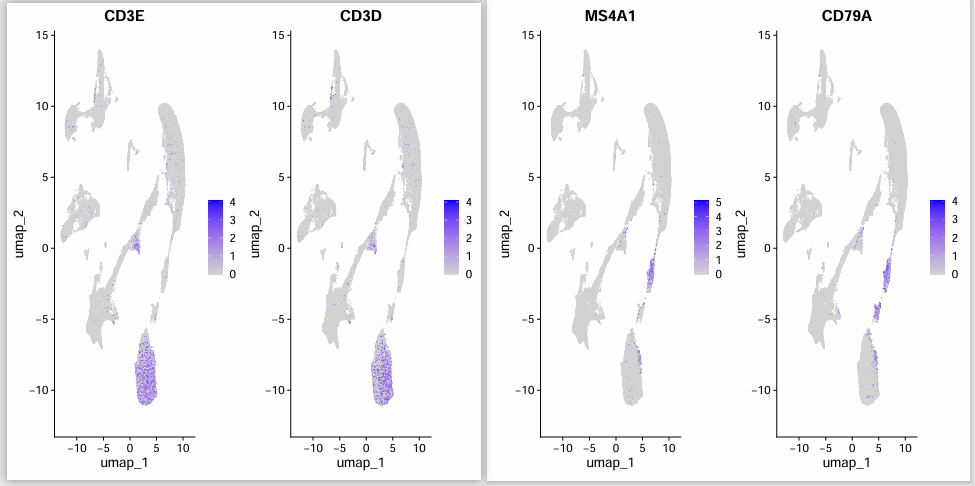
Число гипервариабельных генов – 2000:
Конкретные параметры:
- Гипервариабельный Ген 2000;
- размер ПКА 110;
- Параметры UMAP: n_neighbours — 50, min_dist — 0,1, dims — 1:15.
Видно, что при 3000 гипервариабельных генах Т-клетки и В-клетки разделены. Однако во времена гипермутабельного гена 2000 Т-клетки и В-клетки были связаны друг с другом.
Влияние размерности PCA
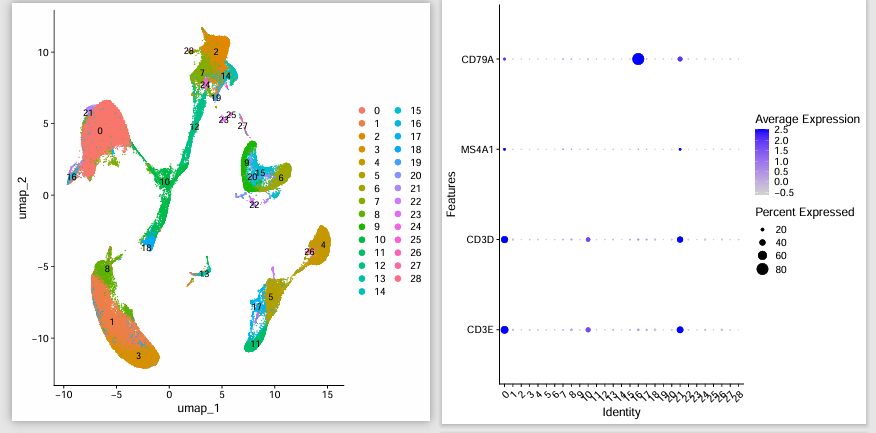
Размерность PCA (npcs) равна 110:
Конкретные параметры:
- 3000 гипервариабельных генов;
- размер ПКА 110;
- Параметры UMAP: n_neighbours — 30, min_dist — 0,3, dims — 1:15.
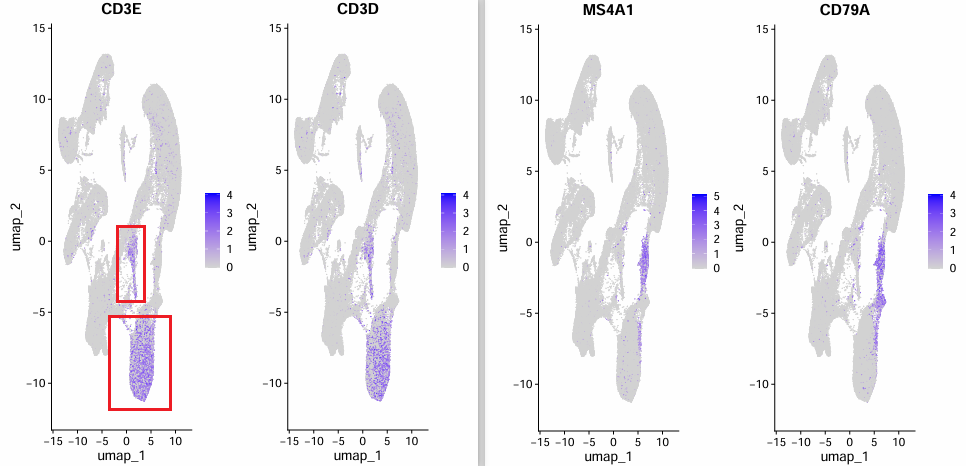
Размерность PCA (npcs) равна 30:
Конкретные параметры:
- 3000 гипервариабельных генов;
- размер ПКА 30;
- Параметры UMAP: n_neighbours — 30, min_dist — 0,3, dims — 1:15.
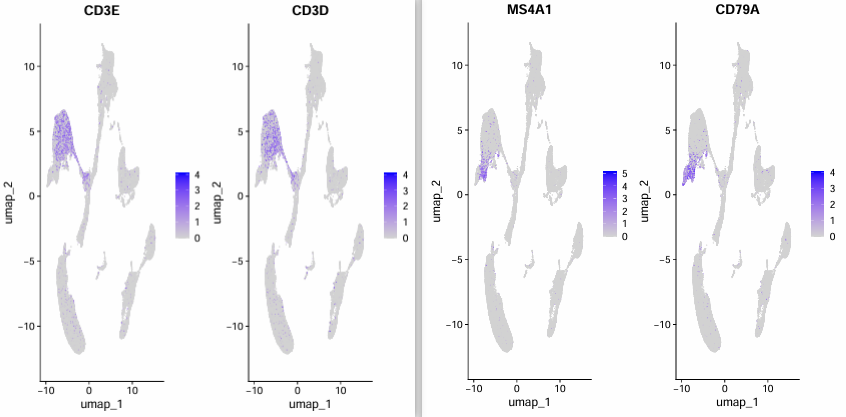
В случае размеров PCA 110 и 30 Т-клетки и В-клетки соединены. Но когда размерность PCA равна 110, Т-клетки делятся на две части, а когда размерность равна 30, Т-клетки составляют одну часть.
Влияние n_neighbours на параметры UMAP
n_neighbours равно 50:
Конкретные параметры:
- 3000 гипервариабельных генов;
- размер ПК 50;
- Параметры UMAP: n_neighbours — 50, min_dist — 0,3, dims — 1:15.
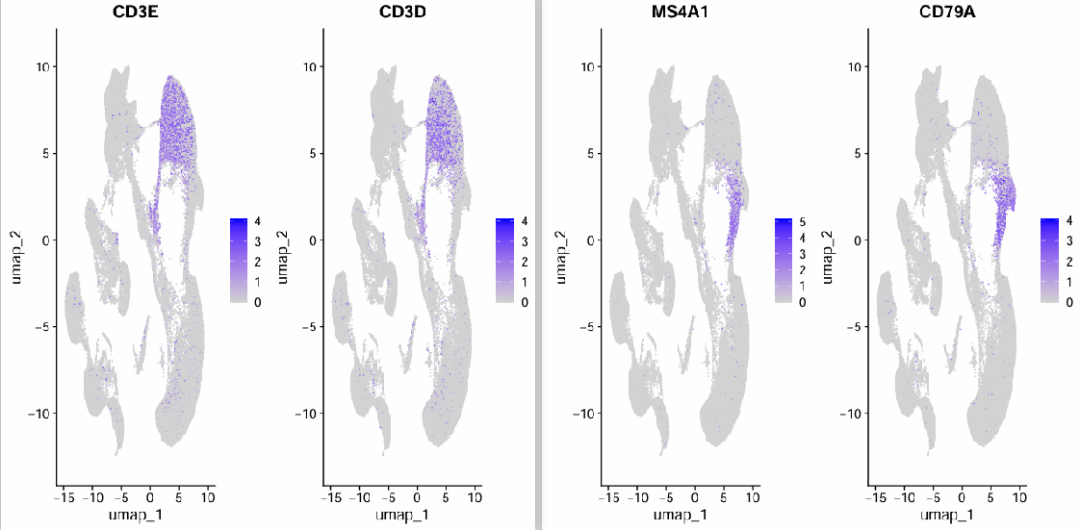
n_neighbours равно 20:
Конкретные параметры:
- 3000 гипервариабельных генов;
- размер ПК 50;
- Параметры UMAP: n_neighbours — 20, min_dist — 0,3, dims — 1:15.
Этот параметр мало влияет
Влияние min_dist на параметры UMAP
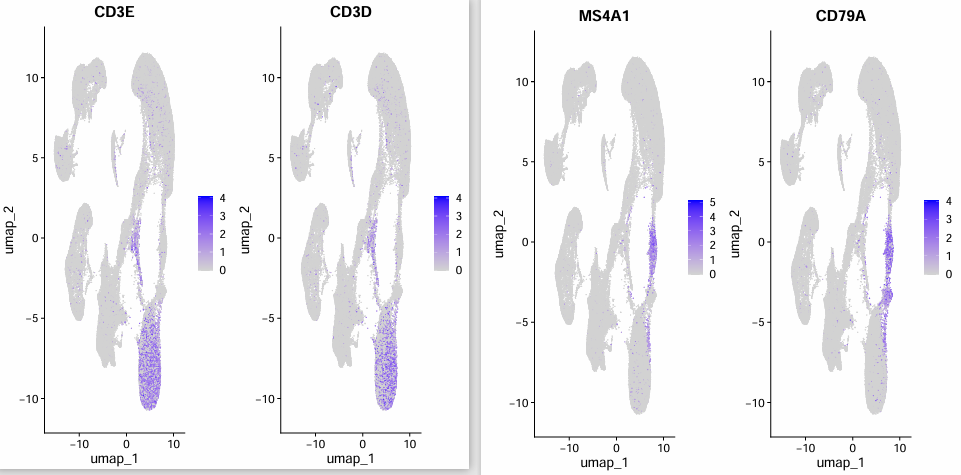
min_dist равен 0,01:
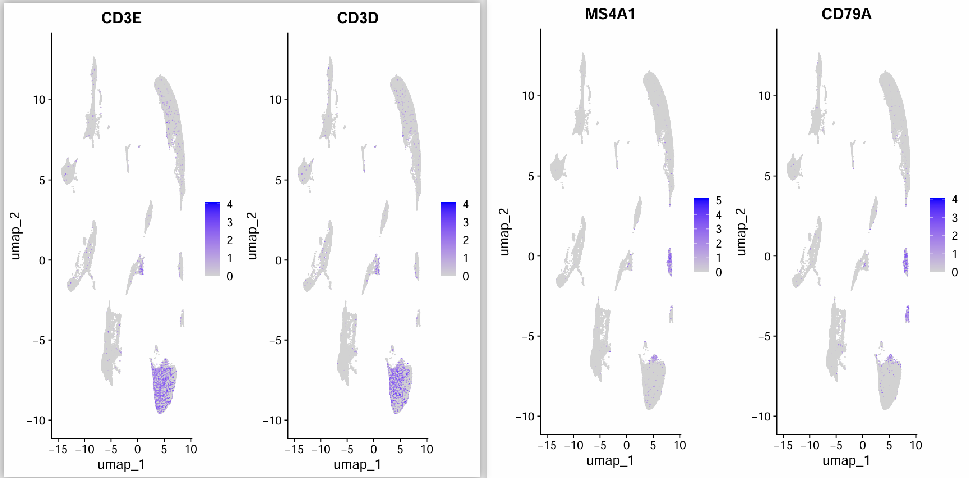
min_dist равен 0,5:
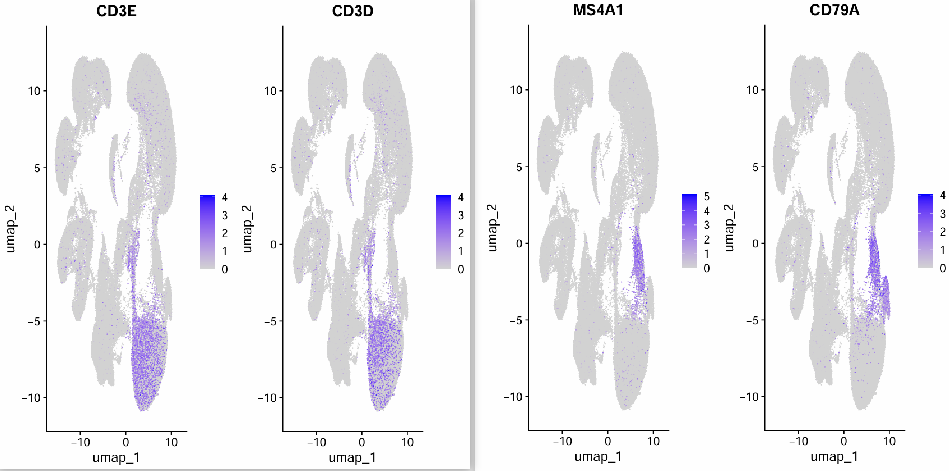
Влияние этого параметра довольно велико. Когда min_dist равен 0,01, Т-клетки и В-клетки могут быть разделены.
Влияние параметров затемнения в UMAP
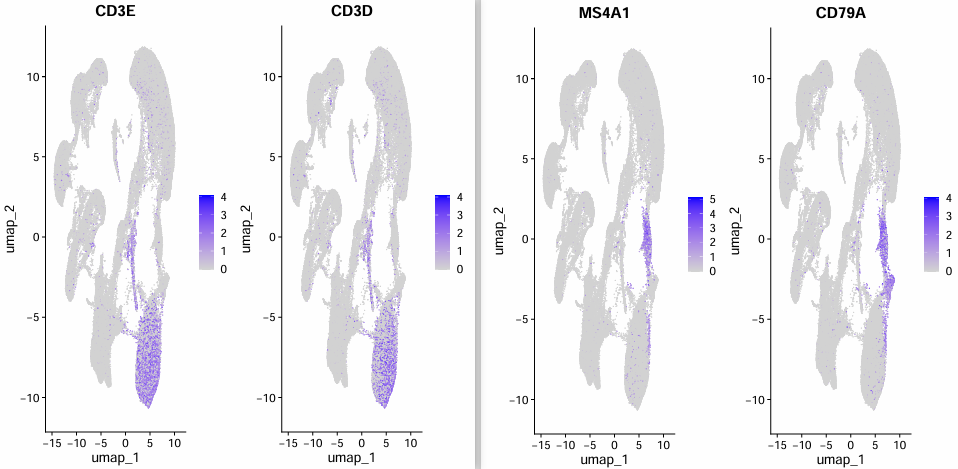
Параметр dims равен 1:27:
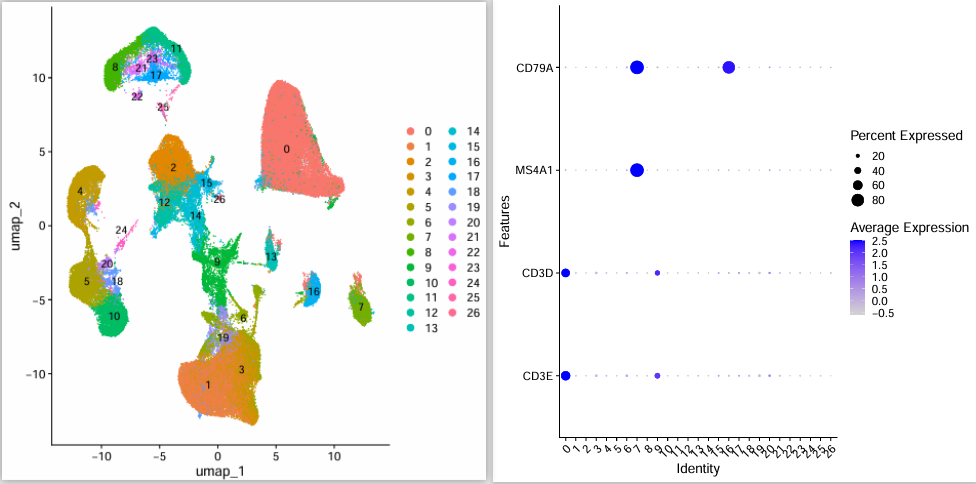
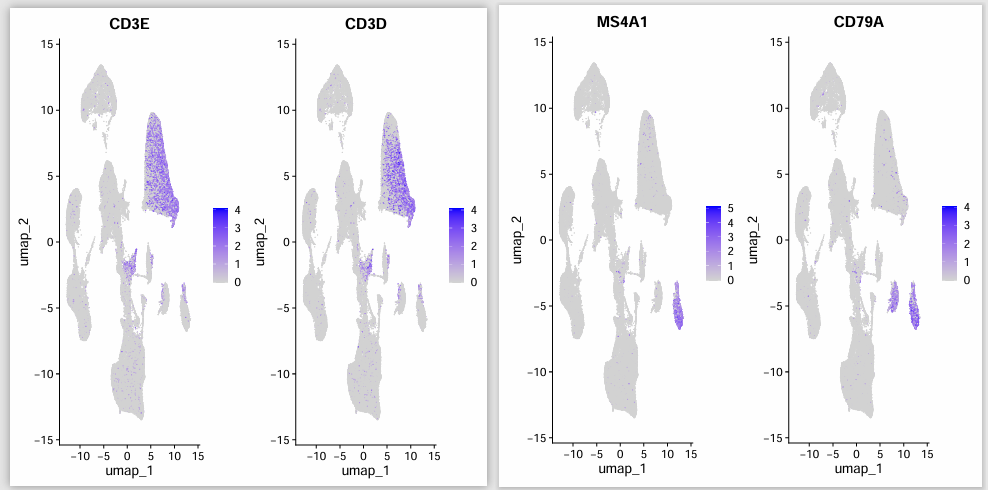
Параметр dims равен 1:15:
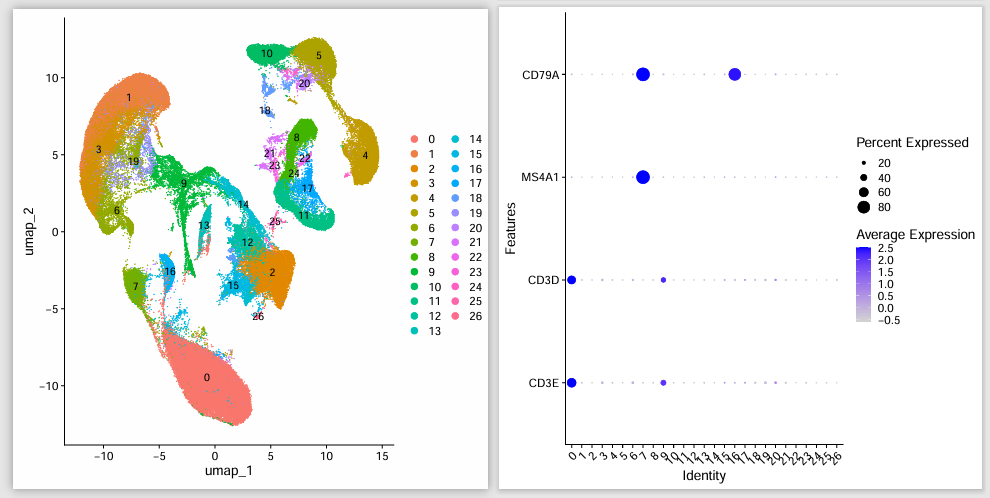
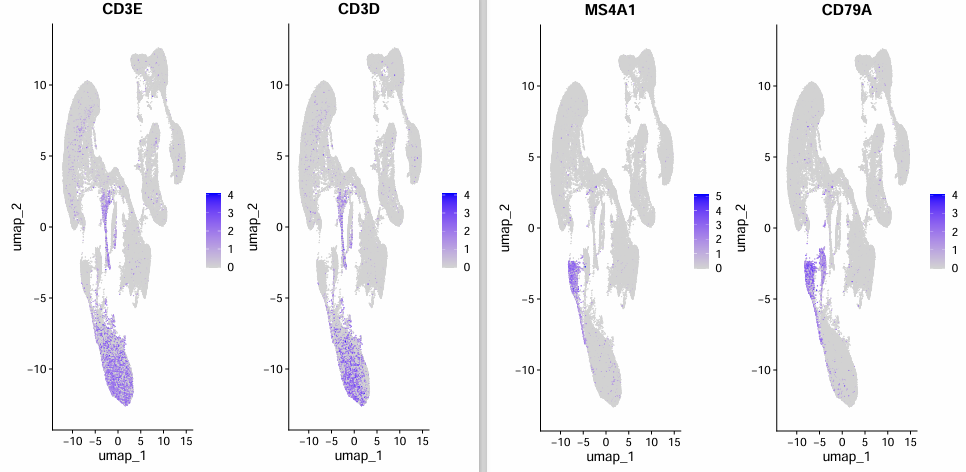
Видно, что когда параметр dims равен 1:27, Т-клетки и В-клетки разделяются. Однако, когда параметр dims равен 1:15, Т-клетки и В-клетки связаны друг с другом.
краткое содержание
Параметры, влияющие на кластеризацию и визуализацию UMAP:
Судя по этому результату, увеличение количества гипервариабельных генов приведет к разделению Т-клеток и В-клеток. Увеличение размера PCA не разделяло Т-клетки и В-клетки, а разделяло Т-клетки на две части.
Ранее в этом твитеПредварительное исследование анализа одиночных ячеек – понимание стандартизации и кластеризации с уменьшением размерности.упомянул:Чем больше число гипервариабельных генов,Чем больше количество ПКиз,Чем больше информации вы сохраните, тем больше информации вы сохраните.,Скорее всего, создаст шум,Он также работает медленнее. Но не слишком мало,В противном случае большая часть данных будет потеряна.
Таким образом, вполне возможно, что увеличение количества гипервариабельных генов позволяет разделять больше различий между Т-клетками и В-клетками. Увеличение размера PCA разделило Т-клетки на две части, возможно, из-за введения шума.
На группировку это не повлияет, только на параметры UMAP-визуализации:
Три параметра n_neighbors, min_dist и dims в параметрах UMAP не влияют на основные атрибуты ячеек, а влияют только на диаграмму визуализации UMAP. Видно, что уменьшение min_dist и увеличение dims приведет к разделению Т-клеток и В-клеток. n_neighbours мало на что влияет.
Я надеюсь, что после прочтения у вас появится общее представление о влиянии изменений параметров на график UMAP.
Прикрепленный пример кода
rm(list=ls())
options(stringsAsFactors = F)
source('../scRNA_scripts/lib.R')
getwd()
###### step1: импортироватьданные ######
# Ссылка для оплаты 800 юань женьминьби
# Ссылка: https://mp.weixin.qq.com/s/tw7lygmGDAbpzMTx57VvFw.
dir='../inputs/'
samples=list.files( dir )
samples = samples[str_detect(samples,"singlecell_count_matrix.txt")]
sceList = lapply(samples,function(pro){
# pro=samples[7]
print(pro)
ct <- data.table::fread( file.path(dir,pro),
data.table = F)
ct[1:4,1:4]
rownames(ct)=ct[,1]
ct=ct[,-1]
#ct=t(ct)
sce =CreateSeuratObject(counts = ct ,
project = gsub('_singlecell_count_matrix.txt.gz','',pro) ,
min.cells = 5,
min.features = 300 )
return(sce)
})
do.call(rbind,lapply(sceList, dim))
sce.all=merge(x=sceList[[1]],
y=sceList[ -1 ],
add.cell.ids = samples )
names(sce.all@assays$RNA@layers)
sce.all[["RNA"]]$counts
# Alternate accessor function with the same result
LayerData(sce.all, assay = "RNA", layer = "counts")
sce.all <- JoinLayers(sce.all)
dim(sce.all[["RNA"]]$counts )
as.data.frame(sce.all@assays$RNA$counts[1:10, 1:2])
head(sce.all@meta.data, 10)
table(sce.all$orig.ident)
library(stringr)
sce.all$orig.ident=str_split(colnames(sce.all),'[-_]',simplify = T)[,1]
table(sce.all$orig.ident)
sce.all
# Если для контроля сложности кода и количества строк
# Ссылка на контроль качества может быть опущена.
###### step2: Контроль качества ######
dir.create("./1-QC")
setwd("./1-QC")
# Если фильтрация слишком жесткая, необходимо изменить код фильтрации.
source('../../scRNA_scripts/qc.R')
sce.all.filt = basic_qc(sce.all)
print(dim(sce.all))
print(dim(sce.all.filt))
setwd('../')
sp='human'
getwd()
sce.all.filt <- NormalizeData(sce.all.filt,
normalization.method = "LogNormalize",
scale.factor = 1e4)
## Оцените влияние параметров
variable_nums <- c(2000,3000,4000)
npcs_nums <- c(30,50,70,90,110)
neighbors_nums <- c(20,30,40,50)
min_dist_nums <- c(0.001,0.01,0.1,0.3,0.5)
# https://zhuanlan.zhihu.com/p/352461768
# RunUMAP: n.neighbors default is 30[5-50]; min.dist default is 0.3[0.001-0.5]
if(F){
variable_nums=2000
npcs_nums=50
neighbors_nums=30
min_dist_nums=0.001
}
for (i in variable_nums){
for (j in npcs_nums){
tmp_sce <- FindVariableFeatures(sce.all.filt, selection.method = "vst",
nfeatures = i)
tmp_sce <- ScaleData(tmp_sce)
tmp_sce <- RunPCA(tmp_sce, features = VariableFeatures(object = tmp_sce),
verbose = FALSE, npcs= j)
tmp_sce <- RunHarmony(tmp_sce, "orig.ident")
tmp_sce <- FindNeighbors(tmp_sce, dims = 1:10, verbose = FALSE,reduction = "harmony")
tmp_sce <- FindClusters(tmp_sce, resolution = 0.5, verbose = FALSE)
table(tmp_sce$seurat_clusters)
for (n in neighbors_nums){
for (d in min_dist_nums){
tmp_sce <- RunUMAP(tmp_sce, dims = 1:15, umap.method = "uwot", metric = "cosine", reduction = "harmony", n.neighbors=n, min.dist=d)
pdf(paste0("pca_object_v",i,".pca_",j,"neighbors_",n,"dist_",d,".pdf"))
p1 <- DimPlot(tmp_sce,label = T,reduction = "umap",raster=F)
print(p1)
genes_to_check = c("CD3E","CD3D","MS4A1","CD79A")
T_genes = c("CD3E","CD3D")
B_genes = c("MS4A1","CD79A")
p2 <- DotPlot(tmp_sce , features = genes_to_check ) +
coord_flip() +
theme(axis.text.x=element_text(angle=45,hjust = 1))
print(p2)
p3 <- FeaturePlot(object = tmp_sce, features = T_genes,raster=F)
print(p3)
p4 <- FeaturePlot(object = tmp_sce, features = B_genes,raster=F)
print(p4)
dev.off()
}
}
}
}


Углубленный анализ переполнения памяти CUDA: OutOfMemoryError: CUDA не хватает памяти. Попыталась выделить 3,21 Ги Б (GPU 0; всего 8,00 Ги Б).
[Решено] ошибка установки conda. Среда решения: не удалось выполнить первоначальное зависание. Повторная попытка с помощью файла (графическое руководство).
Прочитайте нейросетевую модель Трансформера в одной статье

.ART Теплые зимние предложения уже открыты

Сравнительная таблица описания кодов ошибок Amap

Уведомление о последних правилах Points Mall в декабре 2022 года.

Даже новички могут быстро приступить к работе с легким сервером приложений.
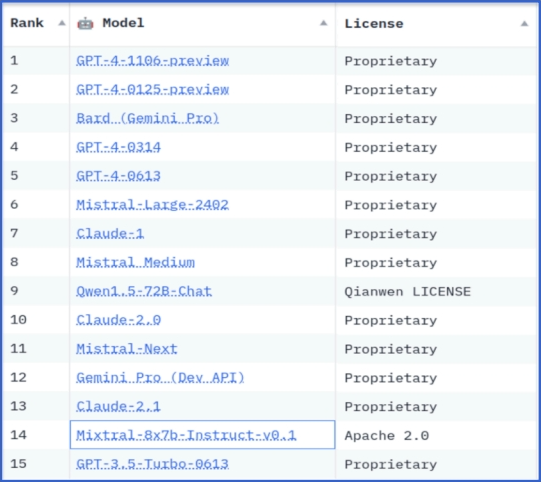
Взгляд на RSAC 2024|Защита конфиденциальности в эпоху больших моделей
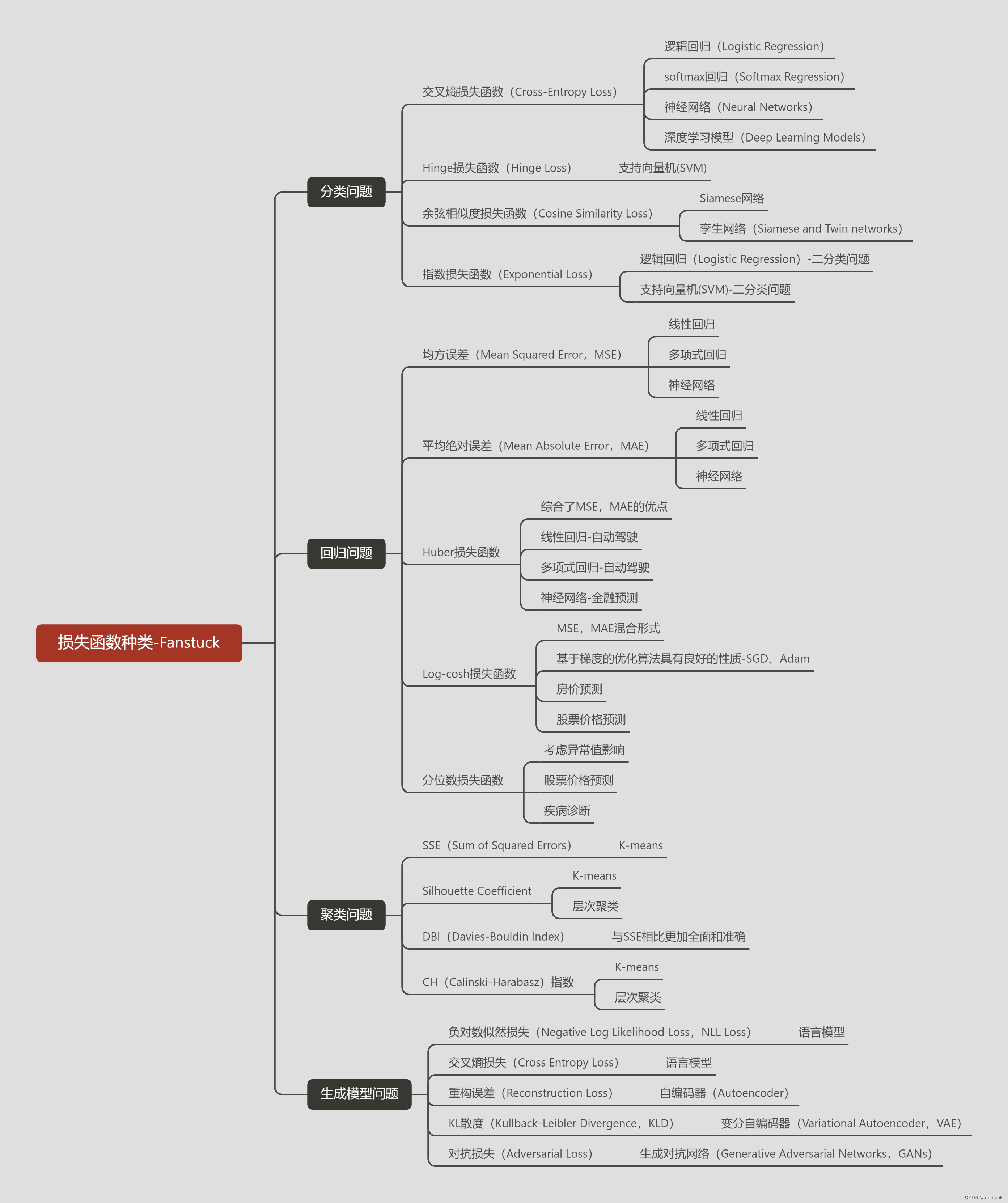
Вы используете ИИ каждый день и до сих пор не знаете, как ИИ дает обратную связь? Одна статья для понимания реализации в коде Python общих функций потерь генеративных моделей + анализ принципов расчета.

Используйте (внутренний) почтовый ящик для образовательных учреждений, чтобы использовать Microsoft Family Bucket (1T дискового пространства на одном диске и версию Office 365 для образовательных учреждений)
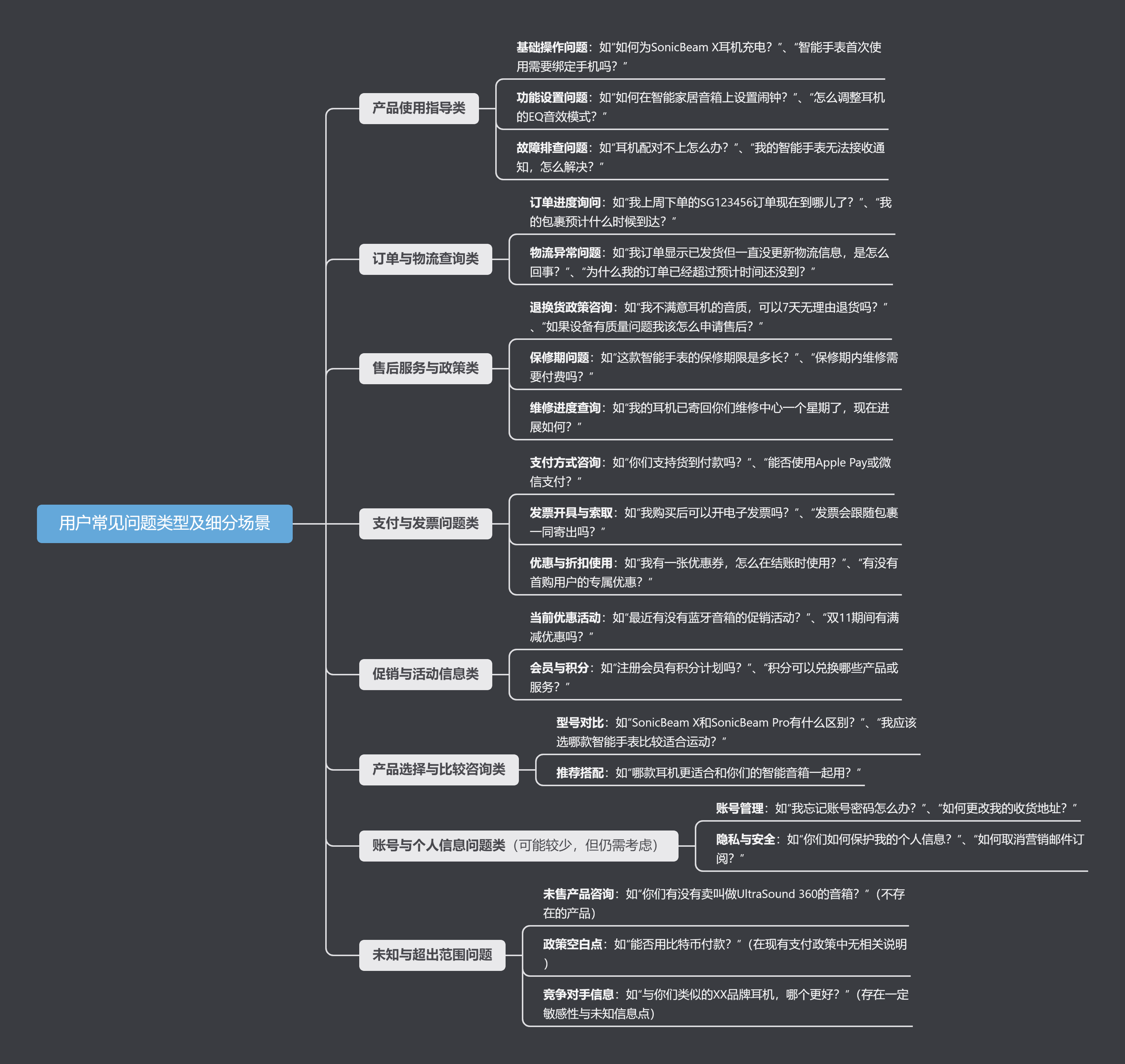
Руководство по началу работы с оперативным проектом (7) Практическое сочетание оперативного письма — оперативного письма на основе интеллектуальной системы вопросов и ответов службы поддержки клиентов

[docker] Версия сервера «Чтение 3» — создайте свою собственную программу чтения веб-текста

Обзор Cloud-init и этапы создания в рамках PVE
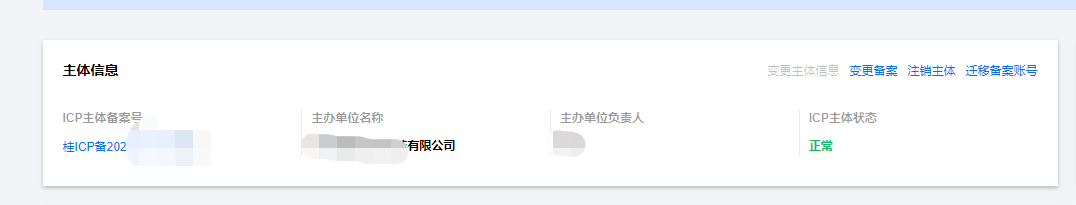
Корпоративные пользователи используют пакет регистрационных ресурсов для регистрации ICP для веб-сайта и активации оплаты WeChat H5 (с кодом платежного узла версии API V3)

Подробное объяснение таких показателей производительности с высоким уровнем параллелизма, как QPS, TPS, RT и пропускная способность.
Удачи в конкурсе Python Essay Challenge, станьте первым, кто испытает новую функцию сообщества [Запускать блоки кода онлайн] и выиграйте множество изысканных подарков!

[Техническая посадка травы] Кровавая рвота и отделка позволяют вам необычным образом ощипывать гусиные перья! Не распространяйте информацию! ! !

[Официальное ограниченное по времени мероприятие] Сейчас ноябрь, напишите и получите приз

Прочтите это в одной статье: Учебник для няни по созданию сервера Huanshou Parlu на базе CVM-сервера.

Cloud Native | Что такое CRD (настраиваемые определения ресурсов) в K8s?

Как использовать Cloudflare CDN для настройки узла (CF самостоятельно выбирает IP) Гонконг, Китай/Азия узел/сводка и рекомендации внутреннего высокоскоростного IP-сегмента

Дополнительные правила вознаграждения амбассадоров акции в марте 2023 г.
Можно ли открыть частный сервер Phantom Beast Palu одним щелчком мыши? Супер простой урок для начинающих! (Прилагается метод обновления сервера)
[Играйте с Phantom Beast Palu] Обновите игровой сервер Phantom Beast Pallu одним щелчком мыши

Maotouhu делится: последний доступный внутри страны адрес склада исходного образа Docker 2024 года (обновлено 1 декабря)

Кодирование Base64 в MultipartFile
5 точек расширения SpringBoot, супер практично!

Глубокое понимание сопоставления индексов Elasticsearch.
15 рекомендуемых платформ разработки с нулевым кодом корпоративного уровня. Всегда найдется та, которая вам понравится.
