Повторное появление набора данных об отдельных клетках рака желудка GSE163558 (2): стандартный процесс Seurat V5
Делиться — это отношение

Предисловие
Привет, друзья, я маленький ученик дерева навыков Шэнсинь «Я не ем яичные желтки». Сегодня вторая фаза серии данных о рецидивах одноклеточного рака желудка GSE163558. В первом выпуске мы загрузили и прочитали данные и успешно создали объект Сёра. В этом выпуске мы будем следовать стандартному процессу Seurat V5, основанному на первом выпуске, проводить контроль качества объектов Seurat, использовать гармоническую интеграцию для пакетной обработки, а также выполнять уменьшение размерности и кластеризацию в соответствии со стандартным процессом.
1Контроль качества
Целью контроля качества (КК) является удаление клеток низкого качества. Клетки низкого качества образуют уникальные субпопуляции, что усложняет группировку результатов. В процессе анализа основных компонентов будут фиксироваться первые несколько основных компонентов, а не различия в качестве; биологические различия, тем самым уменьшая эффект уменьшения размерности. Таким образом, клетки низкого качества могут привести к ошибочным результатам при последующих анализах. Чтобы избежать описанной выше ситуации, нам необходимо исключить эти некачественные клетки перед началом последующего анализа.
Основными показателями КК являются nCount_RNA (количество UMI на клетку), nFeature_RNA (количество генов, обнаруженных в каждой клетке) и «percent_mito» (указатель доли митохондриальных генов в клетке). Кроме того, также могут быть включены два показателя «percent_ribo» (соотношение генов рибосом) и «percent_hb» (соотношение генов эритроцитов).
Количество молекул UMI и генов клетки отражает качество клетки. Если их количество слишком мало, это может быть клеточный мусор; если число слишком велико, это может быть скопление двух или более клеток с высоким содержанием; экспрессия митохондриальных генов может находиться в состоянии апоптоза или клеток в лизированном состоянии с высокой экспрессией рибосомальных генов; Клетки, когда в клетках происходит деградация РНК; клетки с высокой экспрессией генов гемоглобина обычно представляют собой эритроциты, а сами эритроциты не содержат ядер и несут мало генов. Гены, которые они несут, не имеют тесного отношения к заболеваниям и процессам биологического развития. ., поэтому его можно устранить напрямую.
Здесь нам нужно использовать функцию PercentageFeatureSet для расчета «percent_mito», «percent_ribo» и «percent_hb» каждой ячейки соответственно. Сначала загрузите пакет R и импортируйте данные Сёра, сгенерированные за первый период:
getwd()
setwd("")
rm(list=ls())
options(stringsAsFactors = F)
library(Seurat)
library(ggplot2)
library(clustree)
library(cowplot)
library(data.table)
library(dplyr)
sce.all <- readRDS("GSE163558.rds")
Затем начните рассчитывать соотношение митохондриальных генов:
#Рассчитать соотношение митохондриальных генов
mito_genes=rownames(sce.all)[grep("^MT-", rownames(sce.all),ignore.case = T)]
print(mito_genes) #Может быть, 13 митохондриальных генов, ген данных мыши назван строчными буквами "^mt-"
#sce.all=PercentageFeatureSet(sce.all, "^MT-", col.name = "percent_mito")
sce.all=PercentageFeatureSet(sce.all, features = mito_genes, col.name = "percent_mito")
fivenum(sce.all@meta.data$percent_mito)
Рассчитать соотношение генов рибосом
#Рассчитать соотношение генов рибосом
ribo_genes=rownames(sce.all)[grep("^Rp[sl]", rownames(sce.all),ignore.case = T)]
print(ribo_genes)
sce.all=PercentageFeatureSet(sce.all, features = ribo_genes, col.name = "percent_ribo")
fivenum(sce.all@meta.data$percent_ribo)
Затем рассчитайте соотношение генов эритроцитов.
#Рассчитать соотношение генов эритроцитов
Hb_genes=rownames(sce.all)[grep("^Hb[^(p)]", rownames(sce.all),ignore.case = T)]
печать (Hb_genes)
sce.all=PercentageFeatureSet(sce.all, Features = Hb_genes,col.name = "percent_hb")
Fivenum(sce.all@meta.data$percent_hb)
голова(sce.all@meta.data)Визуализируйте вышеуказанные пропорции ячеек
feats <- c("nFeature_RNA", "nCount_RNA", "percent_mito",
"percent_ribo", "percent_hb")
feats <- c("nFeature_RNA", "nCount_RNA")
p1=VlnPlot(sce.all, group.by = "orig.ident", features = feats, pt.size = 0, ncol = 2) +
NoLegend()
p1
w=length(unique(sce.all$orig.ident))/3+5;w
ggsave(filename="Vlnplot1.pdf",plot=p1,width = w,height = 5)
feats <- c("percent_mito", "percent_ribo", "percent_hb")
p2=VlnPlot(sce.all, group.by = "orig.ident", features = feats, pt.size = 0, ncol = 3, same.y.lims=T) +
scale_y_continuous(breaks=seq(0, 100, 5)) +
NoLegend()
p2
w=length(unique(sce.all$orig.ident))/2+5;w
ggsave(filename="Vlnplot2.pdf",plot=p2,width = w,height = 5)
p3=FeatureScatter(sce.all, "nCount_RNA", "nFeature_RNA", group.by = "orig.ident", pt.size = 0.5)
p3
ggsave(filename="Scatterplot.pdf",plot=p3)
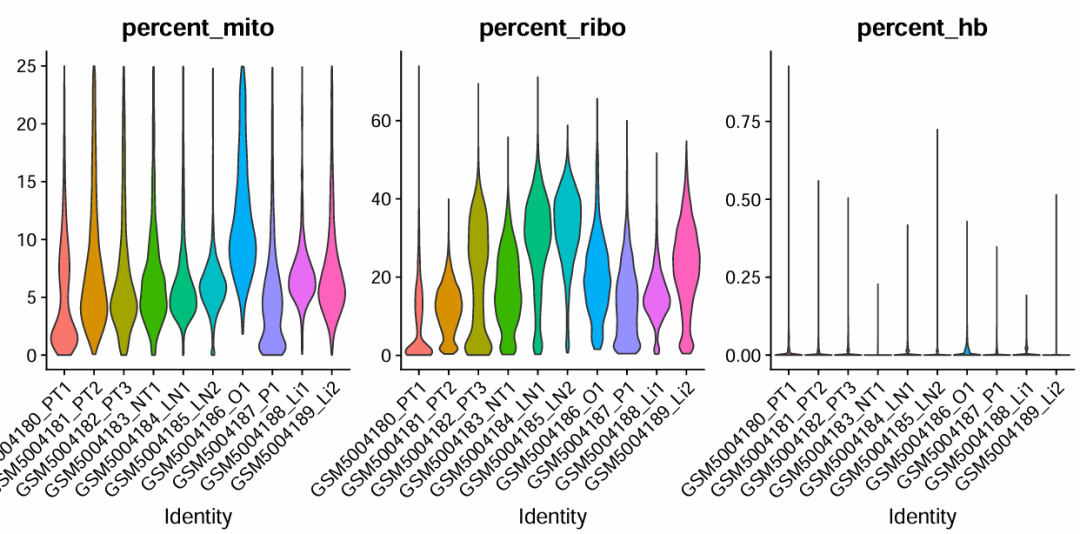
Результат следующий:
p1: nCount_RNA и nFeature_RNA
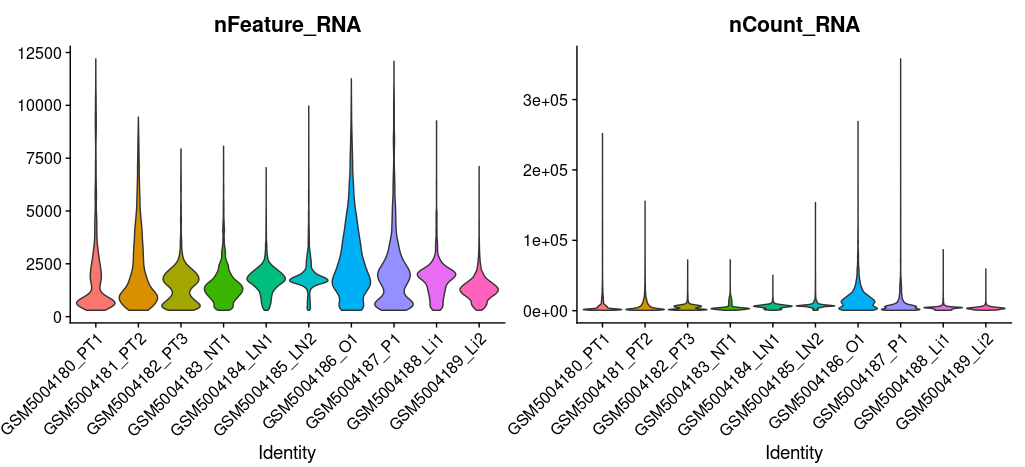
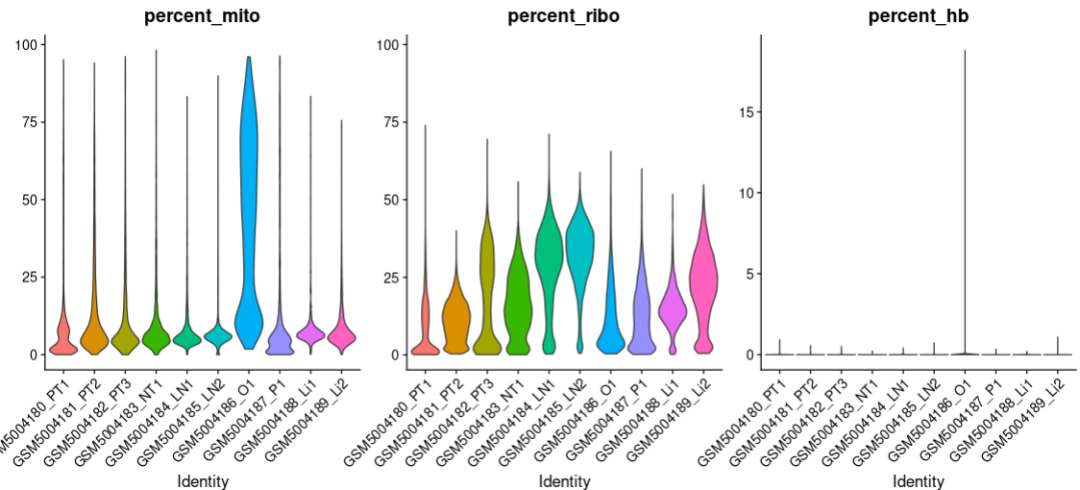
p2: «percent_mito», «percent_ribo» и «percent_hb»
p3: Корреляционный анализ между nCount_RNA и nFeature_RNA
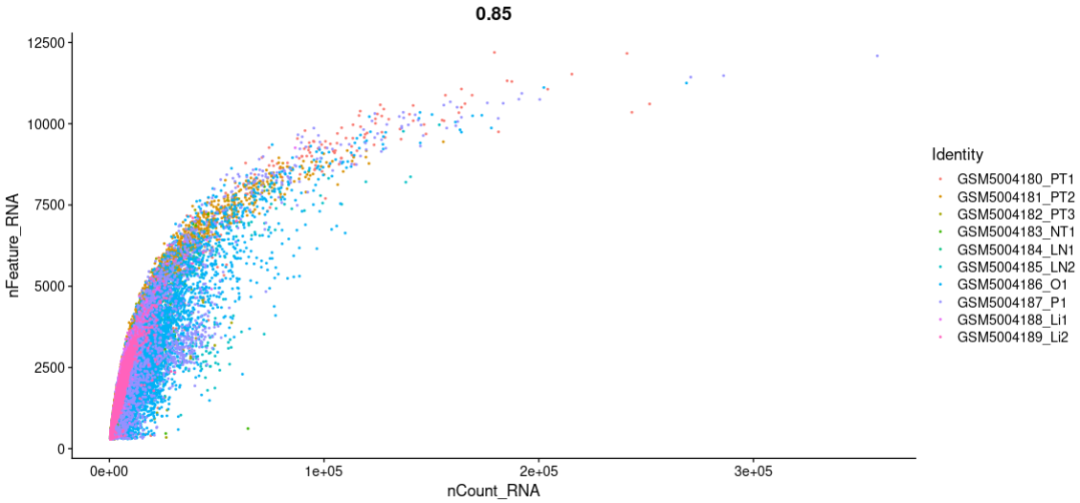
Фильтровать некачественные клетки/гены на основе вышеуказанных показателей
Индекс фильтра 1: клетки с наименьшим количеством экспрессируемых генов и гены с наименьшим количеством экспрессируемых клеток.
Вообще говоря, эта операция фильтрации уже была выполнена при CreateSeuratObject. Если позже вы увидите, что результаты вашей кластеризации с уменьшением размерности одной ячейки странные, вы можете вернуться и посмотреть на процесс контроля качества и сначала просто следовать процессу по умолчанию.
if(F){
selected_c <- WhichCells(sce.all, expression = nFeature_RNA > 500)
selected_f <- rownames(sce.all)[Matrix::rowSums(sce.all@assays$RNA$counts > 0 ) > 3]
sce.all.filt <- subset(sce.all, features = selected_f, cells = selected_c)
dim(sce.all)
dim(sce.all.filt)
}
sce.all.filt = sce.all
# par(mar = c(4, 8, 2, 1))
# С здесь Эта матрица немного великовата, поэтому можно рассматривать случайную выборку
C=subset(sce.all.filt,downsample=100)@assays$RNA$counts
dim(C)
C=Matrix::t(Matrix::t(C)/Matrix::colSums(C)) * 100
most_expressed <- order(apply(C, 1, median), decreasing = T)[50:1]
pdf("TOP50_most_expressed_gene.pdf",width=14)
boxplot(as.matrix(Matrix::t(C[most_expressed, ])),
cex = 0.1, las = 1,
xlab = "% total count per cell",
col = (scales::hue_pal())(50)[50:1],
horizontal = TRUE)
dev.off()
rm(C)
Индикатор фильтра 2: соотношение митохондрий и рибосомальных генов (в соответствии со скрипичной таблицей выше)
Здесь наш критерий фильтрации: процент_мито < 25,percent_ribo > 3,percent_hb < 1. Разные наборы данных и разные организации должны регулировать порог фильтрации в соответствии с конкретными ситуациями.
selected_mito <- WhichCells(sce.all.filt, expression = percent_mito < 25)
selected_ribo <- WhichCells(sce.all.filt, expression = percent_ribo > 3)
selected_hb <- WhichCells(sce.all.filt, expression = percent_hb < 1 )
length(selected_hb)
length(selected_ribo)
length(selected_mito)
sce.all.filt <- subset(sce.all.filt, cells = selected_mito)
sce.all.filt <- subset(sce.all.filt, cells = selected_ribo)
sce.all.filt <- subset(sce.all.filt, cells = selected_hb)
dim(sce.all.filt)
table(sce.all.filt$orig.ident)
length(sce.all.filt$orig.ident)
Давайте посчитаем количество клеток в каждом образце до и после контроля качества:
Количество клеток в каждом образце перед первым этапом контроля качества:

Количество клеток в каждом образце после контроля качества в этот период:

Видно, что после КК количество клеток изменилось с 53093 до 45548, и фильтрация прошла успешно (особенно образец GSM5004186_O1, многие клетки имеют высокое содержание митохондриальных генов. До фильтрации было 5837 клеток. После отфильтровывания некачественных ячеек, осталось 1909).
Визуализируйте отфильтрованную ситуацию:
feats <- c("nFeature_RNA", "nCount_RNA")
p1_filtered=VlnPlot(sce.all.filt, group.by = "orig.ident", features = feats, pt.size = 0, ncol = 2) +
NoLegend()
w=length(unique(sce.all.filt$orig.ident))/3+5;w
ggsave(filename="Vlnplot1_filtered.pdf",plot=p1_filtered,width = w,height = 5)
feats <- c("percent_mito", "percent_ribo", "percent_hb")
p2_filtered=VlnPlot(sce.all.filt, group.by = "orig.ident", features = feats, pt.size = 0, ncol = 3) +
NoLegend()
w=length(unique(sce.all.filt$orig.ident))/2+5;w
ggsave(filename="Vlnplot2_filtered.pdf",plot=p2_filtered,width = w,height = 5)
p1_filtered
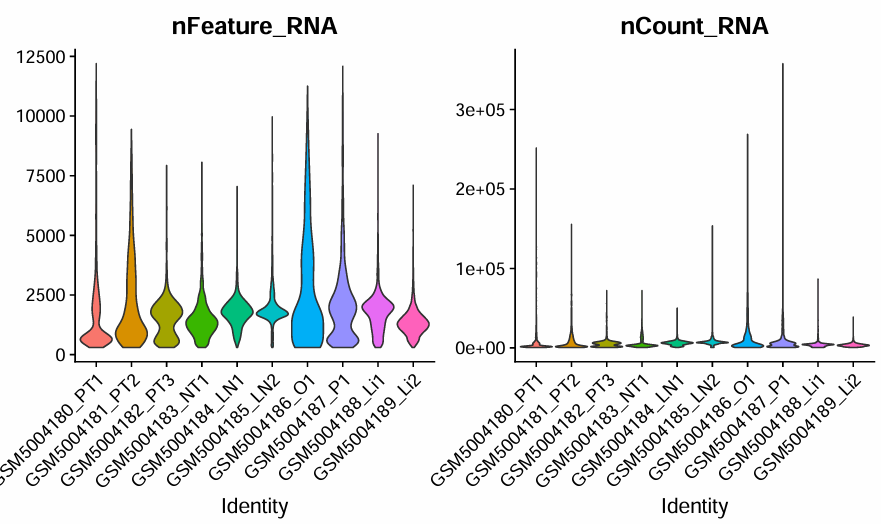
p2_filtered
2 Harmony объединяет несколько образцов отдельных клеток
После выбора ячеек нам необходимо выполнить гармоническое интегрирование. В первом выпуске, когда мы создавали общий объект Seurat, мы использовали функцию слияния, чтобы просто объединить несколько Seurat. merge только объединяет строки и столбцы и не выполняет другую обработку данных.

Прежде чем получить последующую одноклеточную матрицу, образец прошел несколько экспериментальных процессов, и такие переменные, как время, обрабатывающий персонал, реагенты и технологические платформы, станут мешающими факторами. Смешение вышеуказанных факторов приведет к тому, что данные будут иметь групповой эффект. Чтобы избежать, насколько это возможно, мешающих факторов, мы можем строго контролировать технический процесс секвенирования, а также нам необходимо выполнить исправление после события (алгоритм разделения партии) в последующем анализе. В настоящее время широко используемые алгоритмы пакетной обработки для секвенирования отдельных клеток включают Harmony, CCA, RPCA, FastMNN, scVI и т. д. Здесь мы используем Harmony для демонстрации.
Прежде чем Гармония пойдет в партию, нам необходимо осуществить следующий процесс:
Нормализация данных
Метод нормализации данных заключается в выполнении логарифмического преобразования исходного значения выражения «LogNormalize».,Сделайте его общее распределение более согласованным с нормальным распределением. После логарифмического преобразования,Различные гены или клетки также более сопоставимы.,В определенной степени устранить технические различия между разными ячейками.
sce.all.filt <- NormalizeData(sce.all.filt,
normalization.method = "LogNormalize",
scale.factor = 1e4)
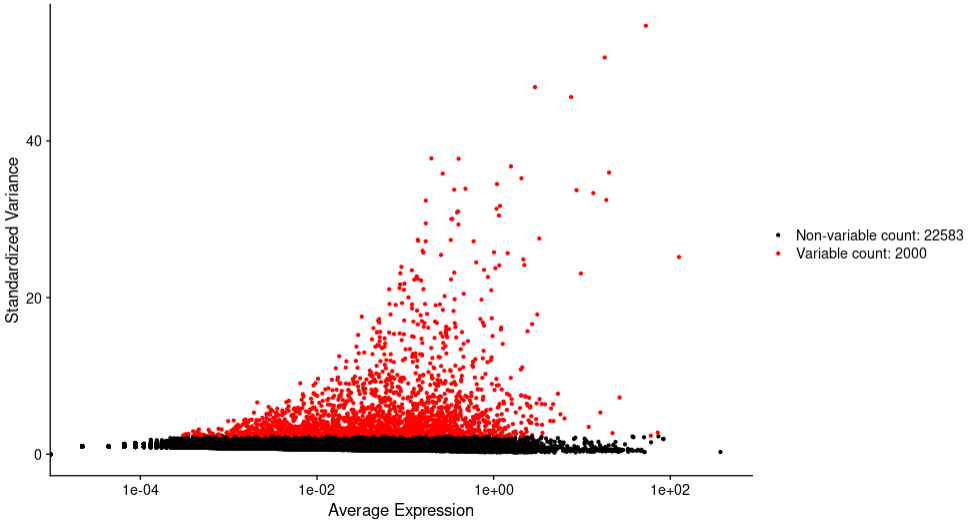
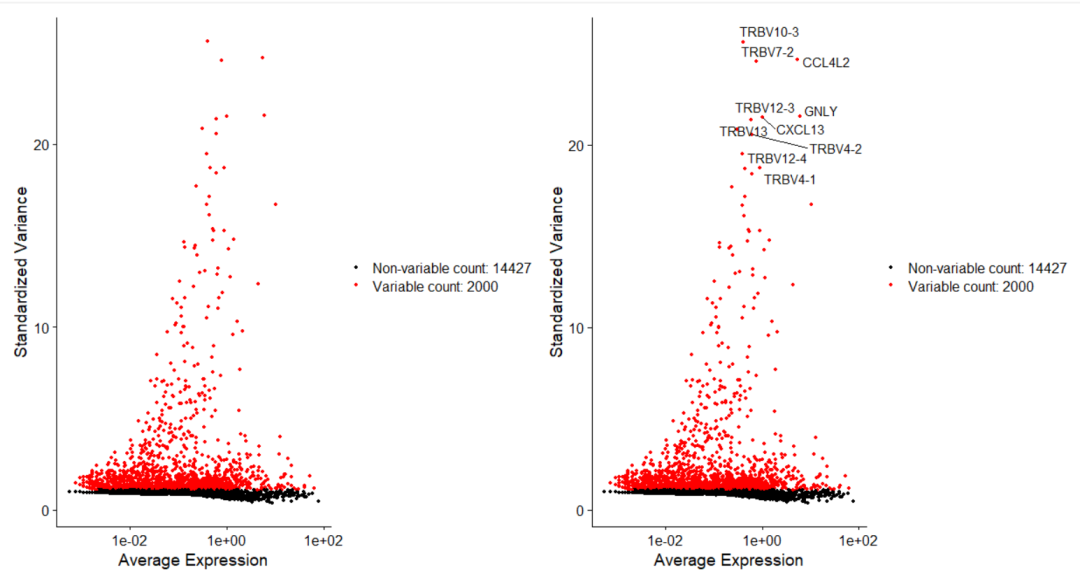
Скрининг гипервариабельных генов
Высоковариабельные гены (HVG) — это гены, экспрессия которых высока в одних клетках и низка в других. По умолчанию на этом этапе фильтруются 2000 HVG для дальнейшего анализа.
sce.all.filt <- FindVariableFeatures(sce.all.filt)
p4 <- VariableFeaturePlot(sce.all.filt)
p4
гипервариабельные гены
Нормализация данных
Нормализация шкалы: измените среднее значение каждого гена во всех клетках на 0 и отметьте дисперсию как 1, придав каждому гену одинаковый вес в последующем анализе, чтобы высокоэкспрессированные гены не доминировали.
sce.all.filt <- ScaleData(sce.all.filt)
Уменьшение линейной размерности PCA
Секвенирование отдельных клеток — это технология высокопроизводительного секвенирования.,Наборы данных, которые он производит, очень многомерны с точки зрения количества клеток и генов. Это сразу указывает на тот факт,т.е. секвенирование одной клеткиданныеполученный"Проклятие измерения"проблемы(Лучшее руководство для отдельных ячеек (4): Уменьшение размерности)。
Понятие «проклятие размерности» было впервые предложено Р. Беллманом. Оно описывает, что теоретически многомерные данные содержат больше информации, но на практике это не так. Данные более высокой размерности часто содержат больше шума и избыточности, поэтому добавление дополнительной информации не способствует последующим этапам анализа.
Чтобы сделать обработку и визуализацию данных более удобными, сохраняя при этом важную информацию о данных, здесь нам необходимо применить PCA (принцип принципа components анализ), а именно технология анализа главных компонент,уменьшатьданные Размеры(Понимание результатов уменьшения размерности одноячеечного PCA)。
sce.all.filt <- RunPCA(sce.all.filt,features = VariableFeatures(object = sce.all.filt))
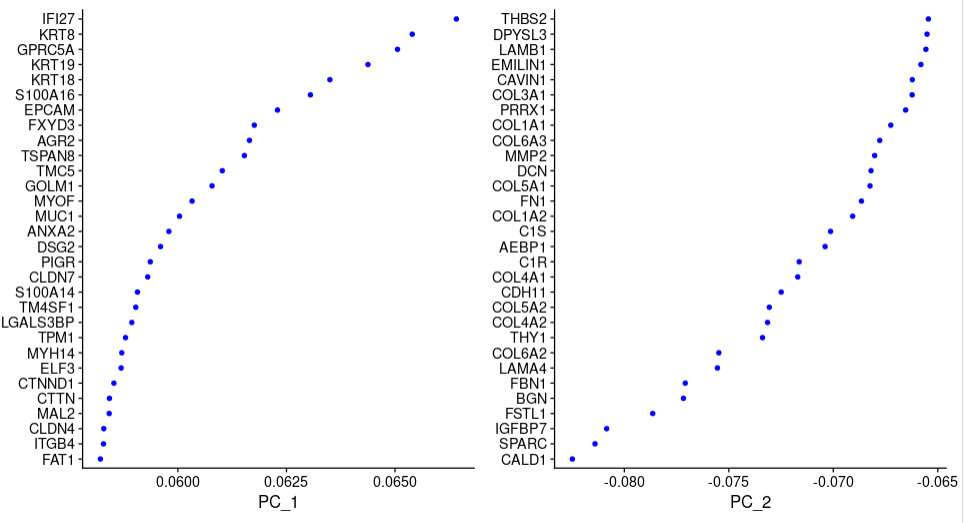
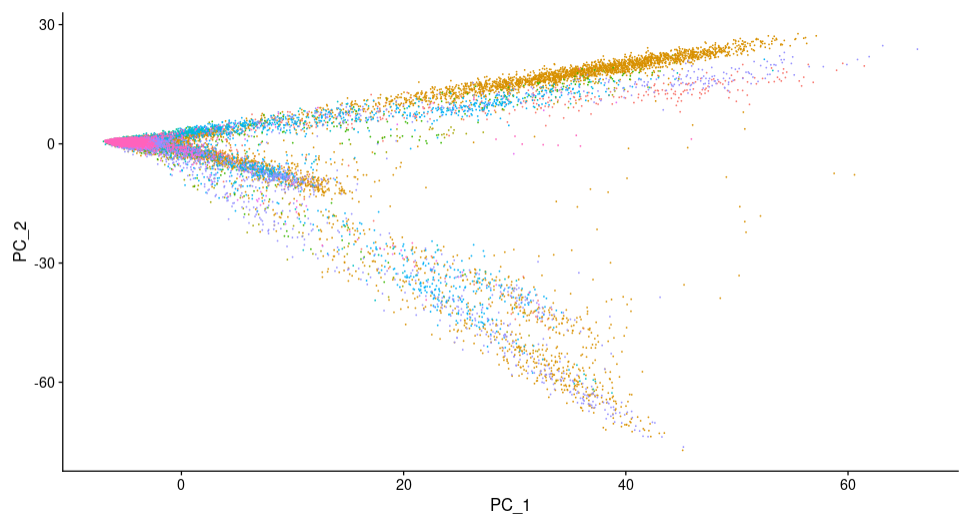
##ВизуализацияPCAрезультат
VizDimLoadings(sce.all.filt, dims = 1:2, reduction = "pca")
DimPlot(sce.all.filt, reduction = "pca") + NoLegend()
DimHeatmap(sce.all.filt, dims = 1:12, cells = 500, balanced = TRUE)
Результаты визуализации PCA
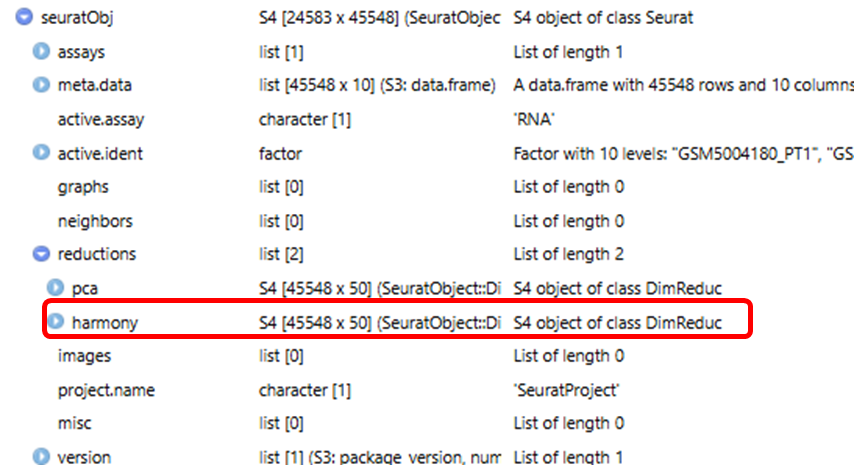
Гармония идет в партию
seuratObj <- RunHarmony(sce.all.filt, "orig.ident")
names(seuratObj@reductions)
[1] "pca" "harmony"
Мы видим, что после RunHarmony результаты Harmony уже заключаются в сокращениях seuratObj.
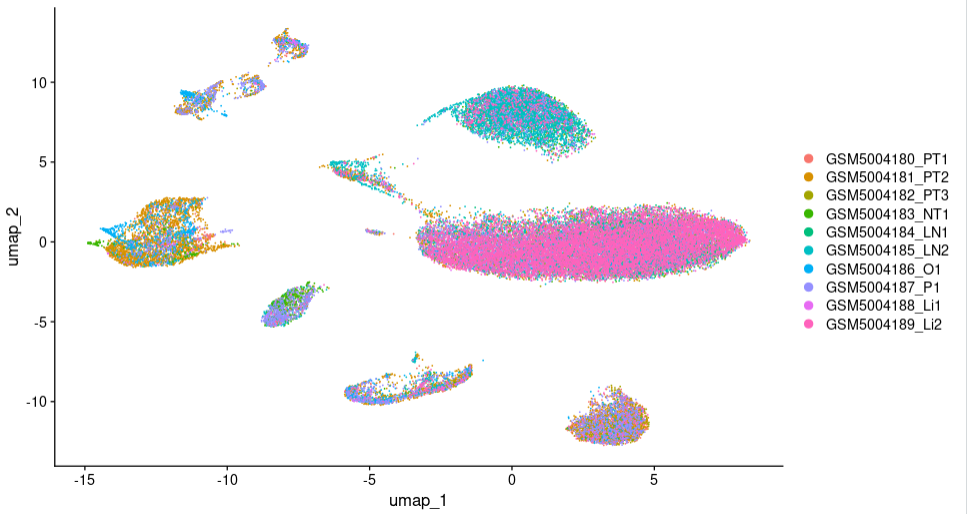
а затем использоватьUMAP/TSNEВизуализация Гармония идет в партию Эффект:
seuratObj <- RunUMAP(seuratObj, dims = 1:15,
reduction = "harmony")
DimPlot(seuratObj,reduction = "umap",label=F )
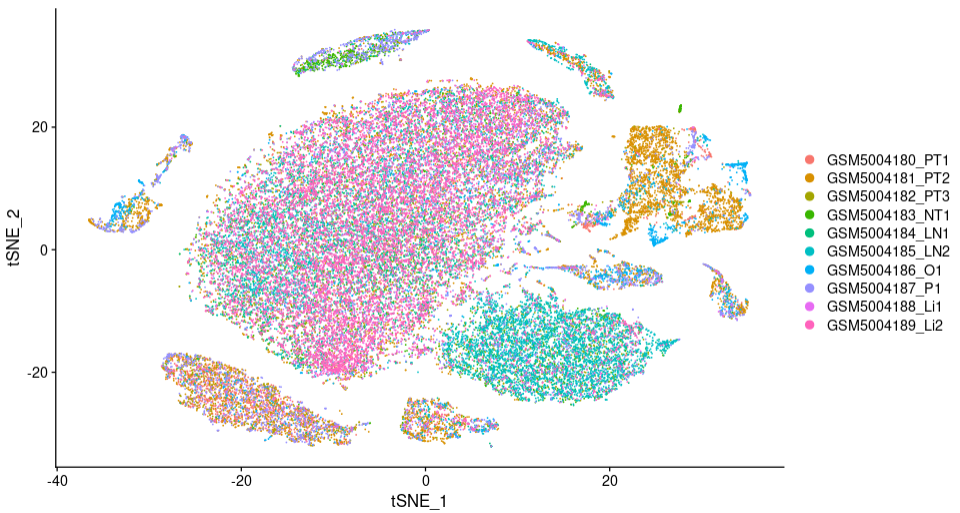
seuratObj <- RunTSNE(seuratObj, dims = 1:15,
reduction = "harmony")
DimPlot(seuratObj,reduction = "tsne",label=F )
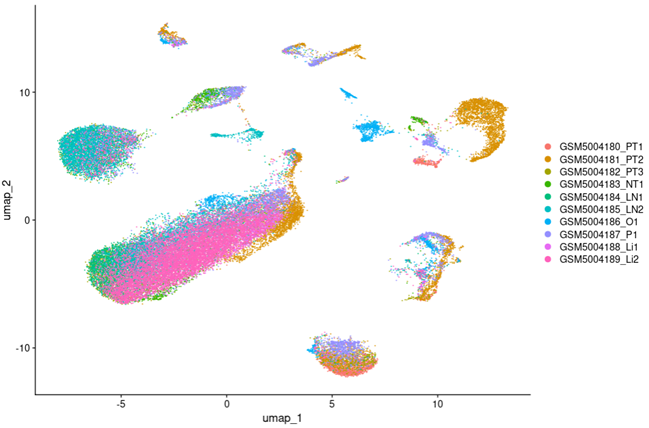
UMAP1
TSNE1
Давайте посмотрим на эффект отсутствия использования Гармония идет в партии:
UMAP2
TSNE2
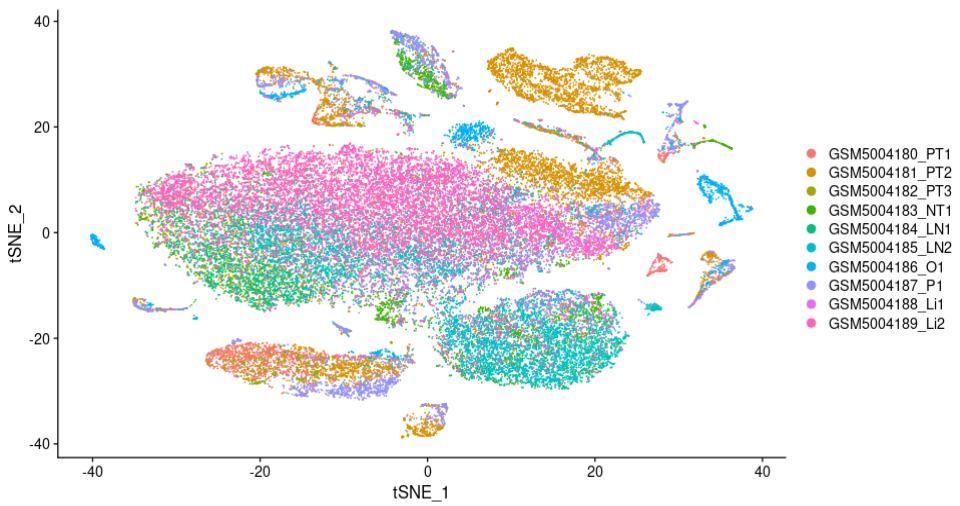
Сравнивая два набора фотографий, мы видим, что,После запуска Гармонии,Точки разброса каждого образца лучше интегрированы,указать, чтоданные Набор необходимо сгруппировать и Гармония идет в партия работает лучше.
3-ячеечная кластеризация
Программное обеспечение Seurat использует алгоритм кластеризации, основанный на теории графов, для кластеризации и группировки ячеек, который включает в себя следующие шаги:
- 1. Постройте отношения кластеризации между ячейками: используйте евклидово расстояние в пространстве PCA, чтобы построить граф отношений кластеризации KNN;
- 2. Весовое значение расстояния отношения кластеризации между ячейками: с использованием подобия Жаккара оптимизация веса ребра между любыми двумя ячейками;
- 3. Кластеризация и группировка: использование Louvain Алгоритм выполняет оптимизацию кластеризации ячеек.
Для определения критериев кластеризации используются две функции FindNeighbors и FindClusters.
Функция FindNeighbors используется для расчета графа ближайших соседей данного набора данных и может возвращать список объектов, содержащих KNN и SNN. Результаты кластеризации также можно оптимизировать с помощью итеративной группировки, чтобы максимизировать стандартную функцию модульности.
Функция FindClusters — это алгоритм кластеризации, основанный на модульной оптимизации общего ближайшего соседа (SNN) для идентификации кластеров ячеек. Важным параметром этой функции является разрешение. Минимальное значение разрешения равно 0, которое делится на 1 категорию; чем больше значение, тем больше кластеров в диапазоне от 0,4 до 1,2. Обычно это дает хорошие результаты для наборов данных из одной ячейки; около 3к.
Давайте сначала запустим FindNeighbors:
sce.all.filt=seuratObj
sce.all.filt <- FindNeighbors(sce.all.filt, reduction = "harmony",
dims = 1:15)
sce.all.filt.all=sce.all.filt
Затем запустите FindClusters. Здесь мы используем цикл for, устанавливаем разные разрешения и наблюдаем эффект кластеризации.
#Установите разные разрешения и наблюдайте за эффектом группировки (какое выбрать?)
for (res in c(0.01, 0.05, 0.1, 0.2, 0.3, 0.5,0.8,1)) {
sce.all.filt.all=FindClusters(sce.all.filt.all, #graph.name = "CCA_snn",
разрешение = разрешение, алгоритм = 1)
}
имена столбцов(sce.all.filt.all@meta.data)
apply(sce.all.filt.all@meta.data[,grep("RNA_snn",colnames(sce.all.filt.all@meta.data))],2,table)
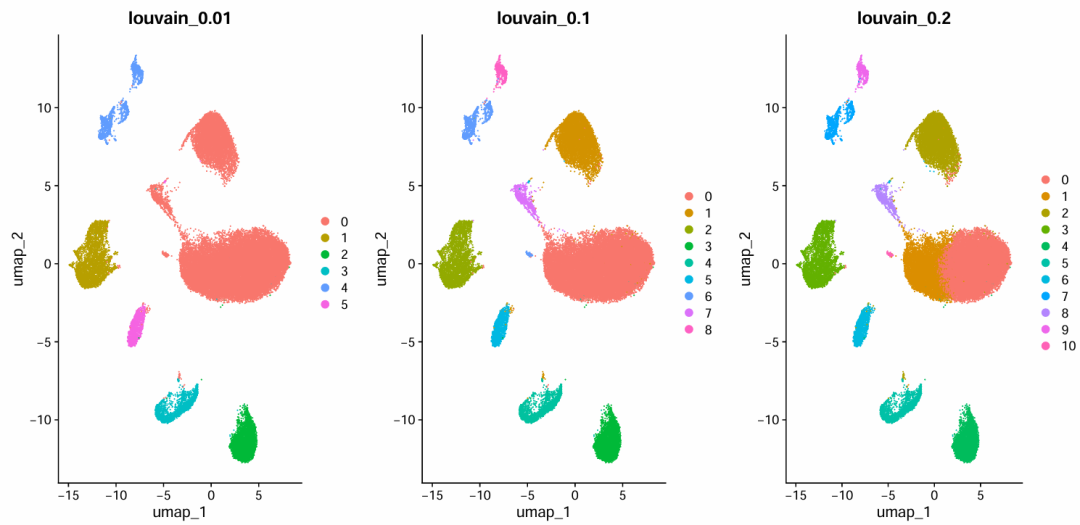
p1_dim=plot_grid(ncol = 3, DimPlot(sce.all.filt.all, Reduction = "umap", group.by = "RNA_snn_res.0.01") +
ggtitle("louvain_0.01"), DimPlot(sce.all.filt.all, Reduction = "umap", group.by = "RNA_snn_res.0.1") +
ggtitle("louvain_0.1"), DimPlot(sce.all.filt.all, Reduction = "umap", group.by = "RNA_snn_res.0.2") +
ggtitle("лувен_0.2"))
ggsave(plot=p1_dim, filename="Dimplot_diff_solve_low.pdf",width = 14)
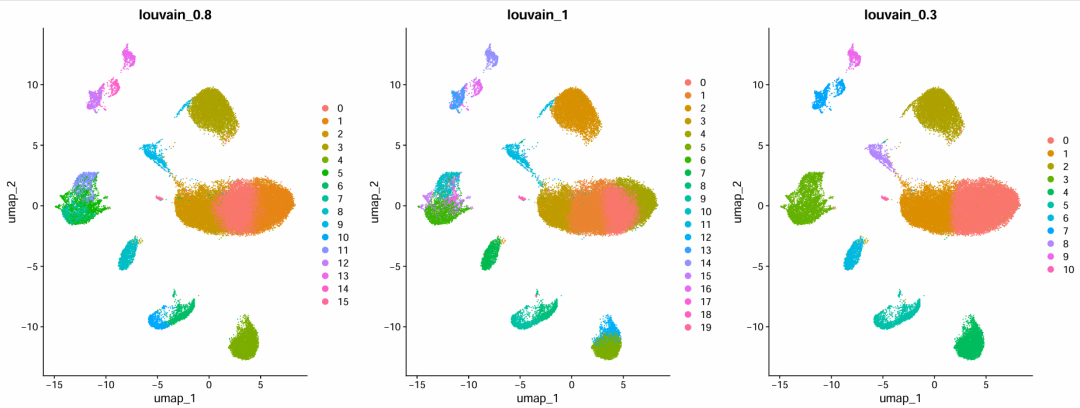
p1_dim=plot_grid(ncol = 3, DimPlot(sce.all.filt.all, Reduction = "umap", group.by = "RNA_snn_res.0.8") +
ggtitle("louvain_0.8"), DimPlot(sce.all.filt.all, сокращение = "umap", group.by = "RNA_snn_res.1") +
ggtitle("louvain_1"), DimPlot(sce.all.filt.all, Reduction = "umap", group.by = "RNA_snn_res.0.3") +
ggtitle("лувен_0.3"))
ggsave(plot=p1_dim, filename="Dimplot_diff_solve_high.pdf",width = 18)
p2_tree=clustree(sce.all.filt.all@meta.data, prefix = "RNA_snn_res.")
ggsave(plot=p2_tree, filename="Tree_diff_solve.pdf")
таблица(sce.all.filt.all@active.ident)Эффект визуальной группировки
Dimplot_diff_resolution_low
Dimplot_diff_resolution_high
Tree_diff_resolution
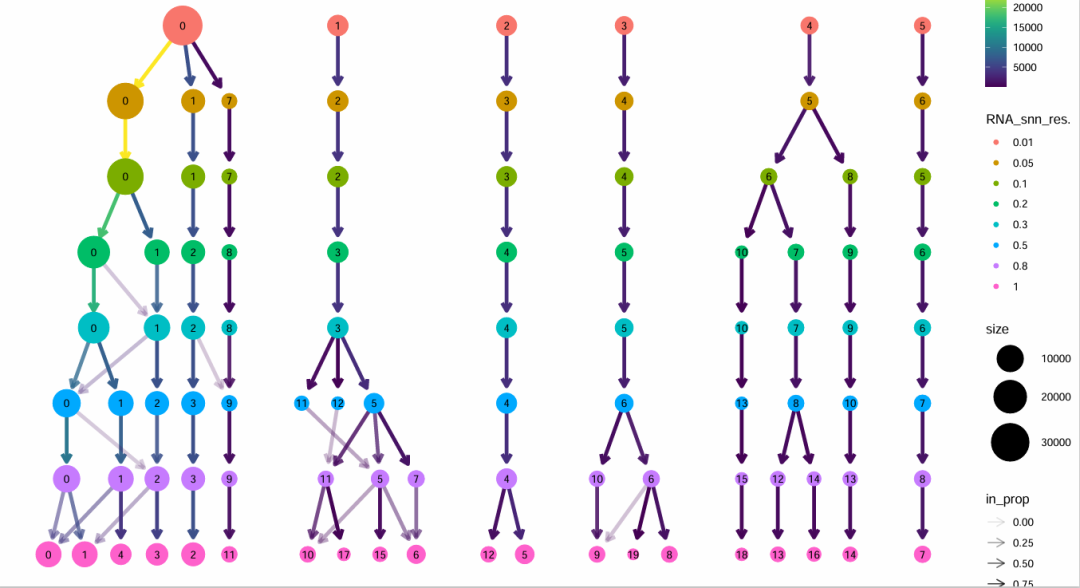
Заключение
В этом выпуске мы представляем стандартный процесс Seurat V5, проводим контроль качества объектов Seurat, используем гармоническую интеграцию для удаления партий и выполняем кластеризацию с уменьшением размерности в соответствии со стандартным процессом. В следующем выпуске мы выберем разрешение = 0,5, чтобы сгруппировать ячейки и аннотировать их на этой основе. Пожалуйста, продолжайте следить за этим, спасибо!


Учебное пособие по Jetpack Compose для начинающих, базовые элементы управления и макет

Код js веб-страницы, фон частицы, код спецэффектов

【новый! Суперподробное】Полное руководство по свойствам компонентов Figma.
🎉Обязательно к прочтению новичкам: полное руководство по написанию мини-программ WeChat с использованием программного обеспечения Cursor.
[Забавный проект Docker] VoceChat — еще одно приложение для мгновенного чата (IM)! Может быть встроен в любую веб-страницу!

Как реализовать переход по странице в HTML (html переходит на указанную страницу)

Как решить проблему зависания и низкой скорости при установке зависимостей с помощью npm. Существуют ли доступные источники npm, которые могут решить эту проблему?

Серия From Zero to Fun: Uni-App WeChat Payment Practice WeChat авторизует вход в систему и украшает страницу заказа, создает интерфейс заказа и инициирует запрос заказа

Серия uni-app: uni.navigateЧтобы передать скачок значения

Апплет WeChat настраивает верхнюю панель навигации и адаптируется к различным моделям.

JS-время конвертации

Обеспечьте бесперебойную работу ChromeDriver 125: советы по решению проблемы chromedriver.exe не найдены

Поле комментария, щелчок мышью, специальные эффекты, js-код
Объект массива перемещения объекта JS

Как открыть разрешение на позиционирование апплета WeChat_Как использовать WeChat для определения местонахождения друзей
Я даю вам два набора из 18 простых в использовании фонов холста Power BI, так что вам больше не придется возиться с цветами!

Получить текущее время в js_Как динамически отображать дату и время в js

Вам необходимо изучить сочетания клавиш vsCode для форматирования и организации кода, чтобы вам больше не приходилось настраивать формат вручную.

У ChatGPT большое обновление. Всего за 45 минут пресс-конференция показывает, что OpenAI сделал еще один шаг вперед.
Copilot облачной разработки — упрощение разработки

Микросборка xChatGPT с низким кодом, создание апплета чат-бота с искусственным интеллектом за пять шагов

CUDA Out of Memory: идеальное решение проблемы нехватки памяти CUDA
Анализ кластеризации отдельных ячеек, который должен освоить каждый&MarkerгенетическийВизуализация

vLLM: мощный инструмент для ускорения вывода ИИ

CodeGeeX: мощный инструмент генерации кода искусственного интеллекта, который можно использовать бесплатно в дополнение к второму пилоту.

Машинное обучение Реальный бой LightGBM + настройка параметров случайного поиска: точность 96,67%
Бесшовная интеграция, мгновенный интеллект [1]: платформа больших моделей Dify-LLM, интеграция без кодирования и встраивание в сторонние системы, более 42 тысяч звезд, чтобы стать свидетелями эксклюзивных интеллектуальных решений.
LM Studio для создания локальных больших моделей
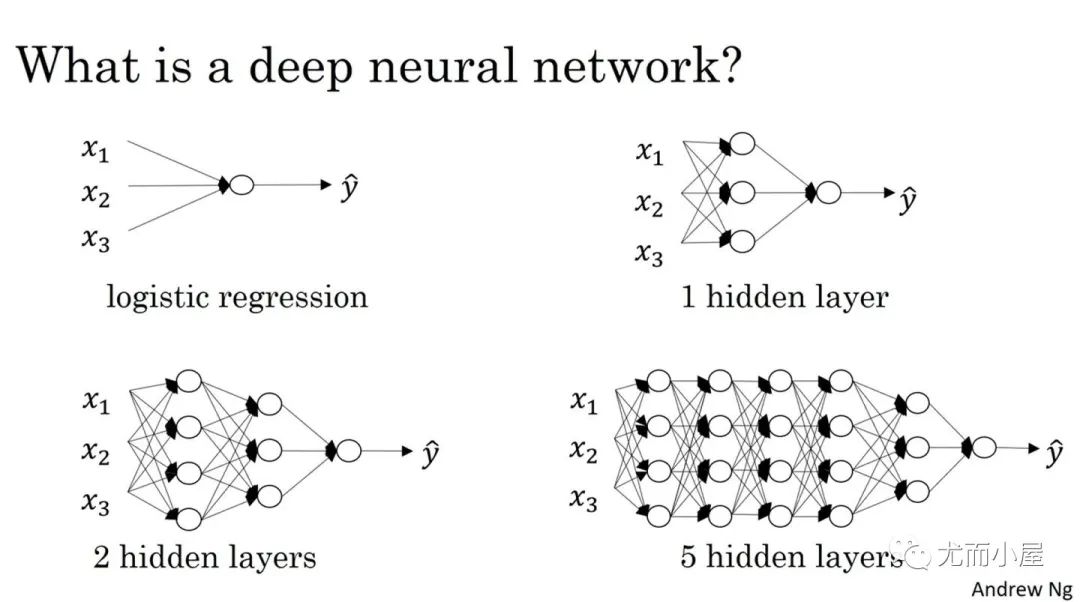
Как определить количество слоев и нейронов скрытых слоев нейронной сети?
