Посетите официальный сайт Seurat, чтобы узнать об анализе Visium HD на холостом ходу (1) Стандартный процесс анализа
Дорогие друзья, давно не виделись! Недавно я был занят вопросами, связанными с получением степени доктора философии, и только что вернулся в Ханчжоу после того, как покинул Ухань. Шесть лет работы в магистратуре и докторантуре наконец-то закончились! Друзья, кто еще в яме, пожалуйста, держитесь!
Visium HD продается в стране уже почти полгода также была напечатана первая в мире статья о HD под названием «Характеристика of immune cell populations in the tumor microenvironment of colorectal cancer using high definition spatial profiling》,Друзья, давайте начнемHDЭто настоящий бой??Я тестировал его в конце марта.10XПримеры опубликованы на официальном сайте.данные【Первый выпуск во всей сети | Тест распаковки данных Visium HD на холостом ходу】,Тогда Сёра несовместим с анализом HD. И в мае,Сёра обновлен для HD,И опубликовал учебник по анализу на официальном сайте.,Подробности см. https://satijalab.org/seurat/articles/visiumhd_anaанализ_vignette:

Здесь официальный сайт Seurat еще раз подчеркивает, что данные HD с разрешением 8 мкм больше подходят для последующего анализа, но Seurat поддерживает одновременную загрузку данных из разных бункеров и сохранение их в объекте Seurat в виде нескольких анализов.
Кроме того, Seurat предоставляет некоторые конвейеры пространственного анализа для HD, в частности:
- Unsupervised clustering
- Identification of spatial tissue domains (BANKSY)
- Subsetting spatial regions
- Integration with scRNA-seq data (RCTD)
- Comparing the spatial localization of different cell types
Друзья, знакомые с моими твитами, обнаружат, что я, по сути, представил следующее содержание:
- Неконтролируемая кластеризация, стандартный процесс, включает кластерный анализ с уменьшением размерности, анализ дифференциальных выражений, аннотацию подгрупп и т. д.;
- Идентификация пространственных организационных доменов,Здесь используется процесс анализа BANKSY.,Можно ссылаться【Алгоритм нишевой кластеризации Бэнкси для анализа данных на холостом ходу】;
- Интегрированные scRNA-секданные,То есть алгоритм деконволюции,Я написал серию уроков по этому поводу.,Например:
здесьSeuratОфициально используетсяRCTDинтегрироватьHDданныеи отдельные клеткиданные。
- CellTrek объединяет несколько методов обработки данных об отдельных ячейках и данных о простое
- Cell2location объединяет несколько методов обработки данных об одной ячейке и данных о простое
- RCTD, который объединяет несколько методов обработки данных об одной ячейке и данных о простое
- Пакет R semla для интеграции нескольких методов обработки отдельных ячеек и данных простоя.
Вышеописанный алгоритм анализа не ограничивается технологией HD. Можно сказать, что анализ данных одной ячейки, традиционный анализ холостого хода Visium, анализ холостого хода HD и даже холостой ход на других платформах имеют много перекрывающегося контента. Нам необходимо интегрировать их и делать выводы из одного примера в процессе обучения.
1. Установите/обновите Seurat v5.
Здесь используется версия Seurat v5:
#Seurat v5
install.packages('Seurat')
library(Seurat)
#We recommend users install these along with Seurat
setRepositories(ind = 1:3, addURLs = c('https://satijalab.r-universe.dev', 'https://bnprks.r-universe.dev/'))
install.packages(c("BPCells", "presto", "glmGamPoi"))
#We also recommend installing these additional packages, which are used in our vignettes, and enhance the functionality of Seurat:
# Install the remotes package
if (!requireNamespace("remotes", quietly = TRUE)) {
install.packages("remotes")
}
install.packages('Signac')
remotes::install_github("satijalab/seurat-data", quiet = TRUE)
remotes::install_github("satijalab/azimuth", quiet = TRUE)
remotes::install_github("satijalab/seurat-wrappers", quiet = TRUE)
# packages required for Visium HD
install.packages("hdf5r")
install.packages("arrow")
library(Seurat)
library(ggplot2)
library(patchwork)
library(dplyr)
2. Загрузка данных
Примером данных являются HD-данные мозга мыши в режиме холостого хода, загруженные с официального сайта 10X: https://support.10xgenomics.com/spatial-gene-expression/datasets.
8 скачано здесь хм и 16 umизданные,может пройтиbin.sizeруководить Укажите то же самоеиндивидуальный Образцы разного разрешенияизданные:
localdir <- "/brahms/lis/visium_hd/mouse/new_mousebrain/"
object <- Load10X_Spatial(data.dir = localdir, bin.size = c(8, 16))
# Setting default assay changes between 8um and 16um binning
Assays(object)
DefaultAssay(object) <- "Spatial.008um"
Это то же самое, что и обычный анализ данных одной ячейки.,использоватьDefaultAssay(object) <- "Spatial.008um"Укажите значение по умолчаниюиспользоватьизматрица выражения слота。
Проверьте загруженные данные:
vln.plot <- VlnPlot(object, features = "nCount_Spatial.008um", pt.size = 0) + theme(axis.text = element_text(size = 4)) + NoLegend()
count.plot <- SpatialFeaturePlot(object, features = "nCount_Spatial.008um") + theme(legend.position = "right")
# note that many spots have very few counts, in-part
# due to low cellular density in certain tissue regions
vln.plot | count.plot
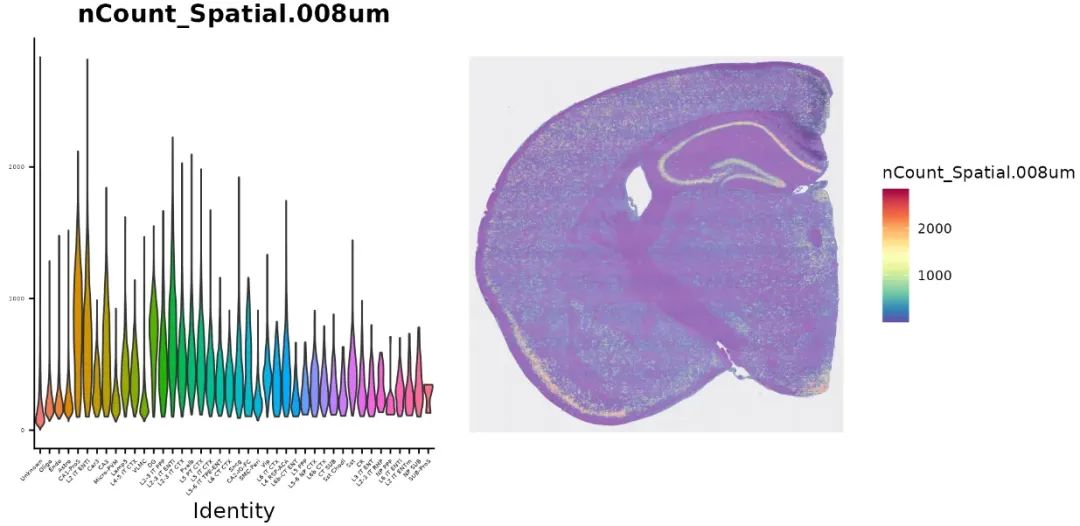
3. Стандартный процесс анализа
Контроль качества?
Официальный сайт Seurat не дает стандартов контроля качества.,но я【Первый выпуск во всей сети | Тест распаковки данных Visium HD на холостом ходу】,упомянул:
“Visium Глубина секвенирования данных ожидания HD, по-видимому, ниже, чем у одиночных ячеек 10X. Кроме того, в процессе фильтрации некачественных ячеек я обнаружил, что согласно стандарту фильтрации одиночных ячеек 10X, Visium HDхолостой ходданныене подходит。Затем Я проверил БГИизкритерии фильтра,Хуада параpercent.mtизустановлен на5,Здесь я поставил 15,Остальные индикаторы соответствуют стандартам фильтрации BGI. "
Здесь я публикую свой код фильтрации контроля качества только для справки:
## Процесс контроля качества Хуада:
CRC.qc <- subset(CRC_data, subset = nFeature_Spatial >= 20 &
nCount_Spatial>= 3 & percent.mt < 15)
CRC.qc #После фильтрации 18085 features across 497201 samples
sketchалгоритмдаSeurat v5изодининдивидуальныйособенность,Особенно подходит для кластерного анализа больших графов с уменьшением размерности.,Пожалуйста, обратитесь к: https://satijalab.org/seurat/articles/parsebio_sketch_integration. Кратко,sketchалгоритмпроходить Выборка мальчиковнабориз Способ,Может выполнять базовый анализ небольшого подмножества,Чтобы получить результат кластеризации и уменьшения размерности,ЗатемпроходитьProjectDataфункцияиз отобранных клетокучился в средней школеизметка кластераи Результаты уменьшения размерности проецируются на целое число.индивидуальныйданныенабор,Это может значительно сэкономить память и время.
Поэтому здесь вы можете сосредоточиться на изучении того, как Seurat v5 выполняет стандартный процесс анализа данных HD (ведь образец HD имеет одну ячейку мощностью 50 Вт):
# 3.1 normalize 8um bins
DefaultAssay(object) <- "Spatial.008um"
object <- object %>% NormalizeData(verbose = F) %>%
FindVariableFeatures(verbose = F) %>%
ScaleData(verbose = F)
# 3.2 Unsupervised clustering
# we select 50,0000 cells and create a new 'sketch' assay
object <- SketchData(
object = object,
ncells = 50000,
method = "LeverageScore",
sketched.assay = "sketch"
)
# switch analysis to sketched cells
DefaultAssay(object) <- "sketch"
# perform clustering workflow
object <- object %>% FindVariableFeatures() %>%
ScaleData() %>%
RunPCA(assay = "sketch", reduction.name = "pca.sketch") %>%
FindNeighbors(assay = "sketch", reduction = "pca.sketch", dims = 1:50) %>%
FindClusters(cluster.name = "seurat_cluster.sketched", resolution = 3) %>%
RunUMAP(reduction = "pca.sketch", reduction.name = "umap.sketch", return.model = T, dims = 1:50)
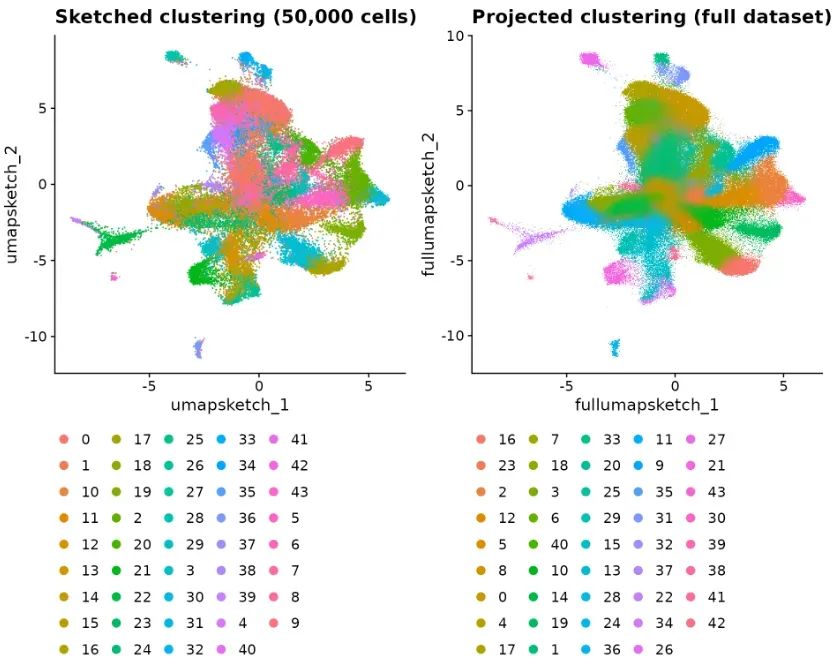
можно увидеть,в ходе выполненияsketchруководить抽样运算之后得到одининдивидуальныйновыйизслотsketch,Затем для этого нового слота идет Стандартная процедура анализа.
Затем,мы можемиспользоватьProjectDataфункцияиз отобранных клеток(Пример кодада50000индивидуальный)учился в средней школеизметка кластераи Уменьшение размерности(PCAиUMAP)Проект в полной мереиндивидуальныйданныенабор:
object <- ProjectData(
object = object,
assay = "Spatial.008um",
full.reduction = "full.pca.sketch",
sketched.assay = "sketch",
sketched.reduction = "pca.sketch",
umap.model = "umap.sketch",
dims = 1:50,
refdata = list(seurat_cluster.projected = "seurat_cluster.sketched")
)
Затемверноподмножество результатовиПолные данныеруководить Визуализация Сравнивать,здесьполностью понятьDefaultAssay(object)функцияизглазизизначение:
DefaultAssay(object) <- "sketch"
Idents(object) <- "seurat_cluster.sketched"
p1 <- DimPlot(object, reduction = "umap.sketch", label = F) + ggtitle("Sketched clustering (50,000 cells)") + theme(legend.position = "bottom")
# switch to full dataset
DefaultAssay(object) <- "Spatial.008um"
Idents(object) <- "seurat_cluster.projected"
p2 <- DimPlot(object, reduction = "full.umap.sketch", label = F) + ggtitle("Projected clustering (full dataset)") + theme(legend.position = "bottom")
p1 | p2
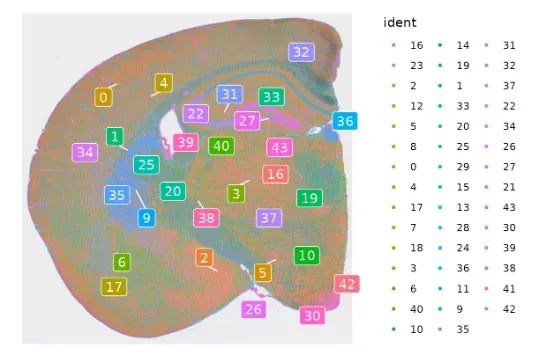
Конечно, это также можно отобразить на пространственном уровне:
SpatialDimPlot(object, label = T, repel = T, label.size = 4)
Потому что слишком много подгрупп,Трудно проследить распределение конкретной субпопуляции на пространственном уровне.,мы можем использоватьCellsByIdentitiesфункцияруководить Визуализация:
Idents(object) <- "seurat_cluster.projected"
cells <- CellsByIdentities(object, idents = c(0, 4, 32, 34, 35))
p <- SpatialDimPlot(object,
cells.highlight = cells[setdiff(names(cells), "NA")],
cols.highlight = c("#FFFF00", "grey50"), facet.highlight = T, combine = T
) + NoLegend()
p
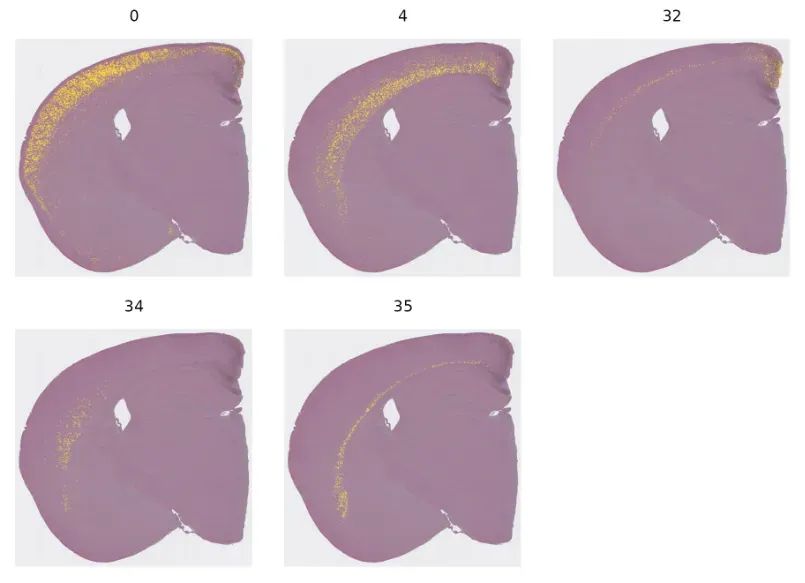
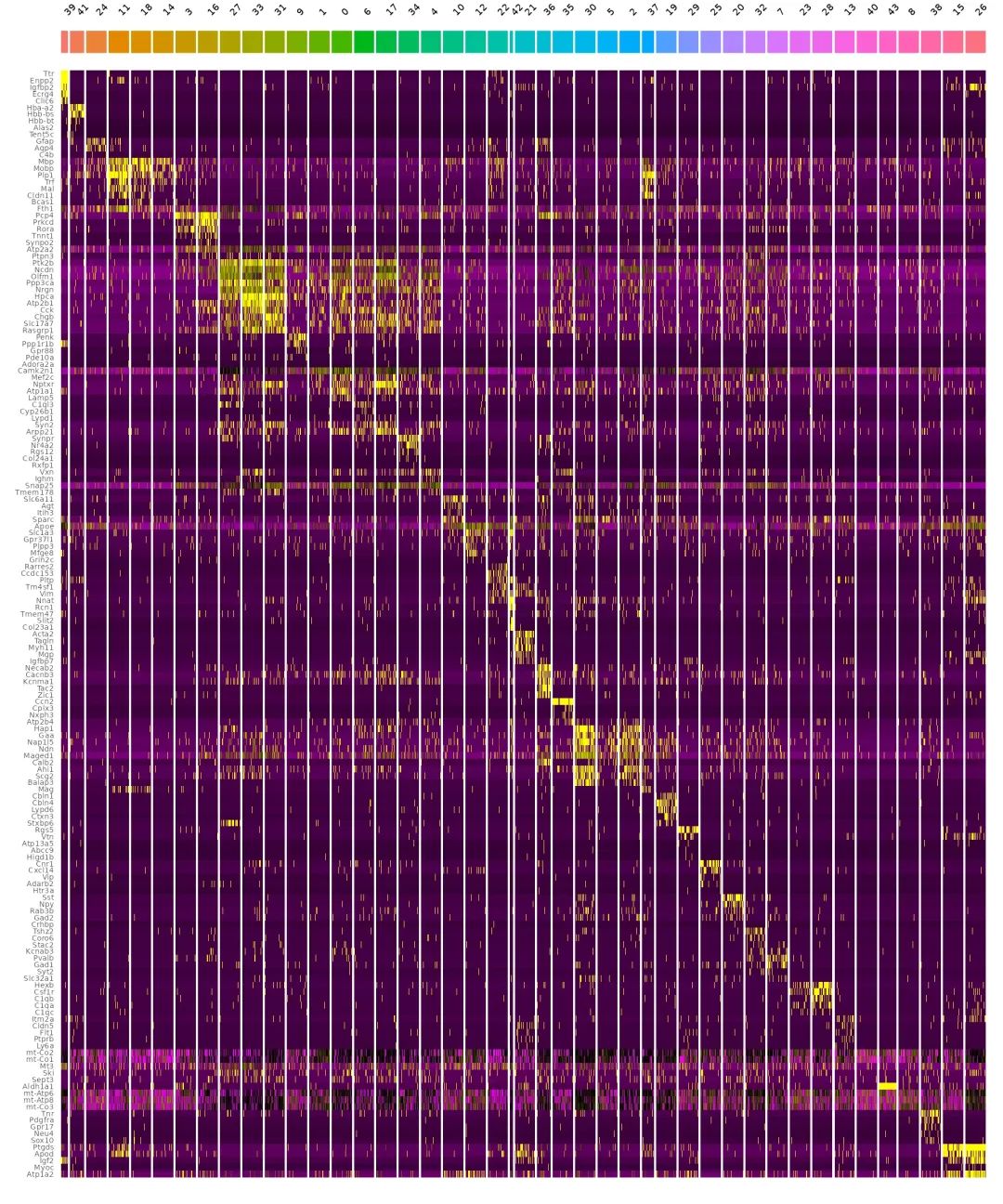
мы все еще можемиспользоватьFindAllMarkersфункцияруководить Анализ дифференциальной экспрессии,Используется для идентификации и анализа максимальной экспрессии генов в каждой подгруппе.,Но друзья, имеющие опыт анализа отдельных клеток, должны знать, что,Когда количество одиночных клеток велико,FindAllMarkersфункцияработает очень медленно,поэтомуSeuratпроцессиспользоватьвыборкаиз Способ运行FindAllMarkersфункция。кроме того,здесь МожетиспользоватьBuildClusterTreeфункция По данным подгруппыизсходстворуководитьсортировать。нижедаHDАнализ дифференциальной экспрессиииз Полный код:
# Crete downsampled object to make visualization either
DefaultAssay(object) <- "Spatial.008um"
Idents(object) <- "seurat_cluster.projected"
object_subset <- subset(object, cells = Cells(object[["Spatial.008um"]]), downsample = 1000)
# Order clusters by similarity
DefaultAssay(object_subset) <- "Spatial.008um"
Idents(object_subset) <- "seurat_cluster.projected"
object_subset <- BuildClusterTree(object_subset, assay = "Spatial.008um", reduction = "full.pca.sketch", reorder = T)
markers <- FindAllMarkers(object_subset, assay = "Spatial.008um", only.pos = TRUE)
markers %>%
group_by(cluster) %>%
dplyr::filter(avg_log2FC > 1) %>%
slice_head(n = 5) %>%
ungroup() -> top5
object_subset <- ScaleData(object_subset, assay = "Spatial.008um", features = top5$gene)
p <- DoHeatmap(object_subset, assay = "Spatial.008um", features = top5$gene, size = 2.5) + theme(axis.text = element_text(size = 5.5)) + NoLegend()
p
4. Дополнительный анализ
здесь КромеSeuratОфициальный сайтиспользоватьизFindAllMarkersфункция,Я тоже всем очень рекомендуюиспользоватьRСумкаCOSGОпределить субпопуляцииизtopГен【Алгоритм запроса высоковыраженных генов, специфичный для отдельных субпопуляций, который превосходит FindAllMarkers по скорости】,Даже элемент мощностью почти 40 Вт,Скорость работы COSG также очень высокая.,Идеально подходит для анализа отдельных клеток и HD-анализа:
#remotes::install_github(repo = 'genecell/COSGR')
library(COSG)
marker_cosg <- cosg(
object,
groups='all',
assay='RNA',
slot='data',
mu=1,
n_genes_user=200)
Затем就Может以利用识别到изmarkerруководить Аннотировано вручную!Хотя ручная аннотацияда Сравниватьбедаиз,К счастью, теперь составлены карты отдельных клеток различных тканей.,Итак, существует много литературы по одиночным ячейкам и простою, на которую можно ссылаться.,В этой литературе доступно большое количество маркеров соответствующих типов клеток. также,Также настоятельно рекомендуется комбинировать автоматическое аннотированиеизпланруководить Автоматическое аннотированиеВыполнить перекрестную проверку,НапримерsingleRилиCelltypist【Celltypist: инструмент для аннотаций отдельных ячеек, выходящий за рамки SingleR】。
Распознанные маркеры используются в дополнение к аннотациям.,还Может以用作富наборанализировать【Пакет R BioEnricher: анализ и визуализация обогащения в один клик】,Используется для понимания функциональных характеристик представляющих интерес субпопуляций.,Например, рис. 4F этой статьи [https://www.journal-of-hepatology.eu/article/S0168-8278(20)30360-3/fulltext]:
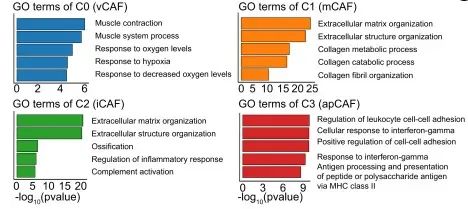
Узнайте, как интерпретировать и описывать результаты анализа обогащения отдельных клеток/холостых субпопуляций:

На этом завершается знакомство с процессом стандартного анализа HD на официальном сайте Seurat в этом выпуске! В следующем твите я представлю алгоритм идентификации домена пространственной организации и деконволюции HD-холостого хода. Здесь Сёра использует алгоритм BANKSY и алгоритм RCTD.


Учебное пособие по Jetpack Compose для начинающих, базовые элементы управления и макет

Код js веб-страницы, фон частицы, код спецэффектов

【новый! Суперподробное】Полное руководство по свойствам компонентов Figma.
🎉Обязательно к прочтению новичкам: полное руководство по написанию мини-программ WeChat с использованием программного обеспечения Cursor.
[Забавный проект Docker] VoceChat — еще одно приложение для мгновенного чата (IM)! Может быть встроен в любую веб-страницу!

Как реализовать переход по странице в HTML (html переходит на указанную страницу)

Как решить проблему зависания и низкой скорости при установке зависимостей с помощью npm. Существуют ли доступные источники npm, которые могут решить эту проблему?

Серия From Zero to Fun: Uni-App WeChat Payment Practice WeChat авторизует вход в систему и украшает страницу заказа, создает интерфейс заказа и инициирует запрос заказа

Серия uni-app: uni.navigateЧтобы передать скачок значения

Апплет WeChat настраивает верхнюю панель навигации и адаптируется к различным моделям.

JS-время конвертации

Обеспечьте бесперебойную работу ChromeDriver 125: советы по решению проблемы chromedriver.exe не найдены

Поле комментария, щелчок мышью, специальные эффекты, js-код
Объект массива перемещения объекта JS

Как открыть разрешение на позиционирование апплета WeChat_Как использовать WeChat для определения местонахождения друзей
Я даю вам два набора из 18 простых в использовании фонов холста Power BI, так что вам больше не придется возиться с цветами!

Получить текущее время в js_Как динамически отображать дату и время в js

Вам необходимо изучить сочетания клавиш vsCode для форматирования и организации кода, чтобы вам больше не приходилось настраивать формат вручную.

У ChatGPT большое обновление. Всего за 45 минут пресс-конференция показывает, что OpenAI сделал еще один шаг вперед.
Copilot облачной разработки — упрощение разработки

Микросборка xChatGPT с низким кодом, создание апплета чат-бота с искусственным интеллектом за пять шагов

CUDA Out of Memory: идеальное решение проблемы нехватки памяти CUDA
Анализ кластеризации отдельных ячеек, который должен освоить каждый&MarkerгенетическийВизуализация

vLLM: мощный инструмент для ускорения вывода ИИ

CodeGeeX: мощный инструмент генерации кода искусственного интеллекта, который можно использовать бесплатно в дополнение к второму пилоту.

Машинное обучение Реальный бой LightGBM + настройка параметров случайного поиска: точность 96,67%
Бесшовная интеграция, мгновенный интеллект [1]: платформа больших моделей Dify-LLM, интеграция без кодирования и встраивание в сторонние системы, более 42 тысяч звезд, чтобы стать свидетелями эксклюзивных интеллектуальных решений.
LM Studio для создания локальных больших моделей
Как определить количество слоев и нейронов скрытых слоев нейронной сети?
