Полный геном – анализ вариаций генома человека (PacBio) (2) – использование CCS
1. Процесс создания геномной библиотеки PacBio SMRTbell
1. Структура библиотеки PacBio SMRTbell
Библиотека секвенирования, созданная платформой секвенирования PacBio, имеет форму гантели. Вот почему это называется SMRT bell, Как показано в правой части рисунка 1. Его основными компонентами являются: разъем в форме шпильки (Шпилька Адаптер) и матрица двухцепочечной ДНК (Double Stranded DNA Шаблон). После того, как текст построен,、Перед секвенированиемЕще нужно завершитьSMRT библиотека колоколов, секвенирование Primer、DNA Работа смешивания полимеразы (отжиг праймера секвенирования связывается с кольцевым адаптером секвенирования, а затем комплекс библиотеки праймер-колокольчик связывается с ДНК-полимеразой). Рисунок 1 справа и рисунок 2 показаны.
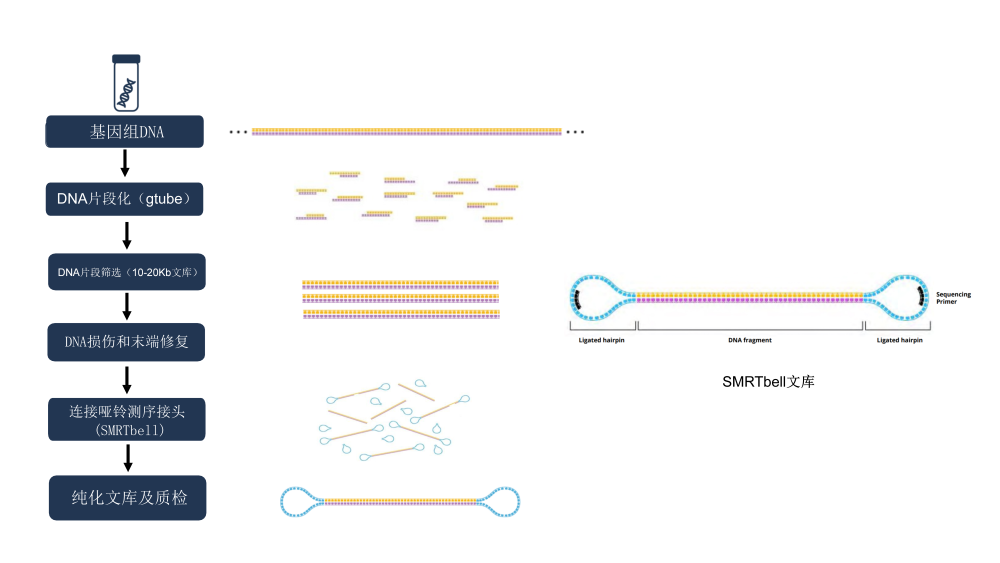
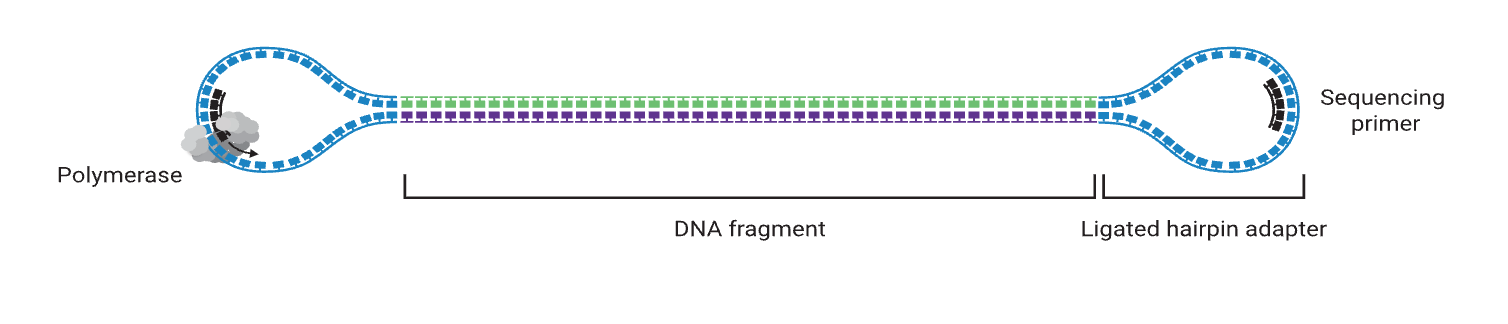
2. Процесс создания геномной библиотеки SMRTbell.
В качестве примера возьмем геномную библиотеку HiFi (библиотека 10–20 КБ), как показано слева на рисунке 1:
1) После получения геномной ДНК (г ДНК) путем экстракции нуклеиновых кислот сначала используйте пробирку G-трубку или систему Megaruptor для фрагментации генома до подходящего размера (обычно 20 КБ для геномов животных и растений для создания библиотеки и 10 КБ для геномов животных и растений). для микробных геномов для создания библиотеки);
2) Получите полную вставку двухцепочечной ДНК, выполнив такие действия, как удаление одноцепочечных выступов, восстановление повреждений и восстановление концов;
3) Создайте библиотеку секвенирования SMRTbell, подключив адаптеры SMRTbell к обоим концам двухцепочечной ДНК для получения кольцевой матрицы.
4) После завершения лигирования адаптера продукт лигирования необходимо очистить и применить ферментную обработку для расщепления линейных или внутренне поврежденных кольцевых молекул ДНК (бесплатный адаптер-шпилька, шаблоны ДНК с адаптерами, не соединенными с обоими концами, кольцевая ДНК с внутренними повреждениями). ) Шаблон), после завершения ферментативной обработки обычно используется система Bulepippin или Sage ELF для разрезания геля и восстановления библиотеки в пределах целевого диапазона размеров.
два、PacBio Subreads and HiFi reads
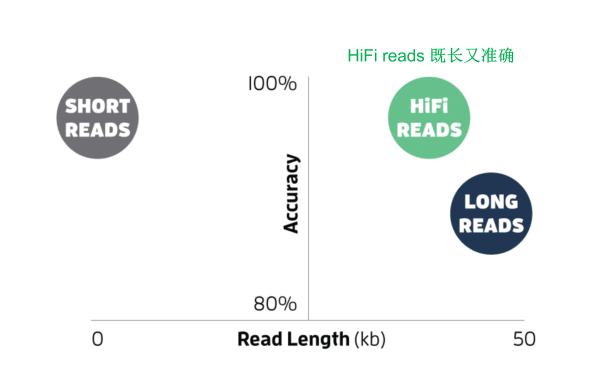
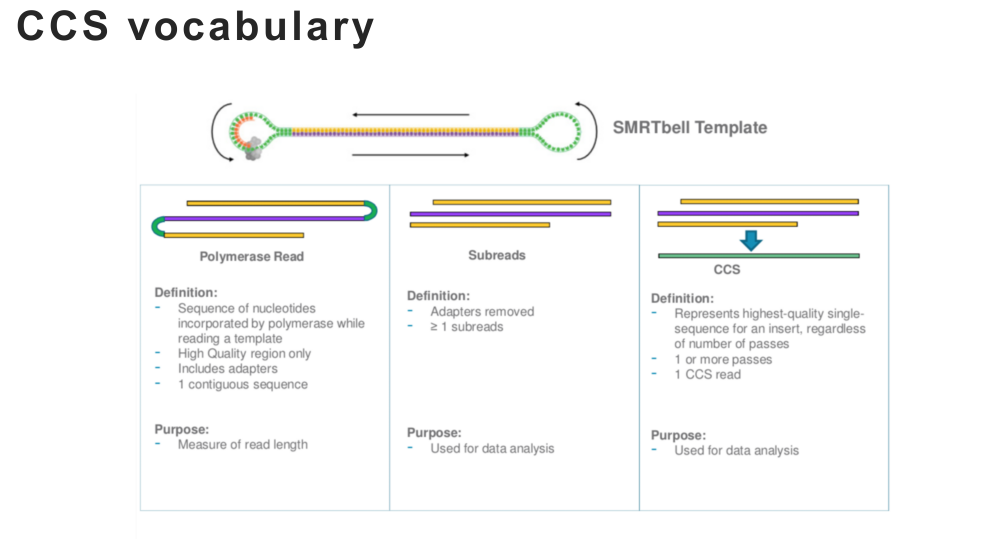
HiFi reads(High Fidelity reads) — это консенсусная последовательность на основе Circular, запущенная PacBio в 2019 году. Consensus Режим секвенирования, CCS) создает систему, учитывающую большую длину считывания (~10-20 kb)Высокая точность(>99%Точность)данные последовательности секвенирования (Рисунок 3).
Для фрагмента ДНК, подлежащего секвенированию, в режиме секвенирования CCS длина считывания фермента (полимеразная read) намного больше длины вставленного фрагмента,Полимераза будет выполнять секвенирование по катящемуся кругу вокруг матрицы ДНК.,Вставленный целевой фрагмент будет секвенирован несколько раз.。Создано за один цикл секвенированияслучайныйОшибки последовательности,Серия избыточныхSubreadsчтобы исправить себя。проходитьPacBioРазработано компаниейалгоритм CCSПосле самостоятельной коррекции,Наконец, очень точныйCCS read, Поскольку качество секвенирования каждой базы выше, оно называется HiFi. read (Рисунок 4).
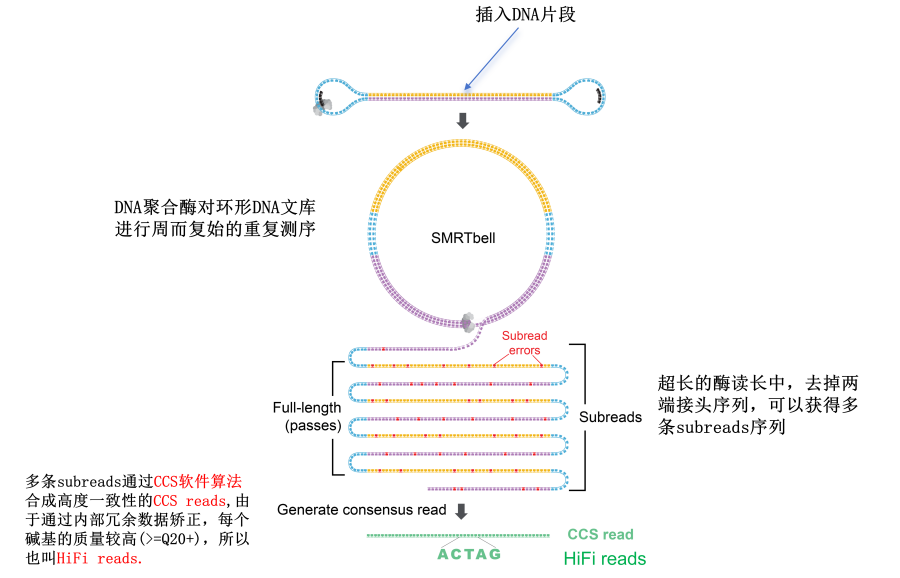
3. PacBio передает данные в HiFi, считывает данные.
Pacbio Sequel II Платформа поддерживает CLR (непрерывный Long Читает)иCCS(Циркуляр Consensus Секвенирование) два метода секвенирования. CLRРежим для библиотек очень длинных фрагментов(> 25 kb), данные субчтений, полученные вне машины, не будут обрабатываться дальше и могут использоваться непосредственно в качестве исходных данных для последующего анализа. Единственным недостатком является то, что точность каждого чтения ниже.
Начиная со второй половины 2022 года, последний комплект сборки библиотеки SMRTbell prep kit 3.0 отказывается от режима CLR и полностью принимает режим построения и секвенирования библиотеки CCS. Таким образом, субчтения, исходящие от машины, должны проходить через алгоритм CCS для устранения избыточности. и конвертировать подчтения в чтения HiFi. Пользователям платформы Pacbio Sequel II данные дополнительного чтения вне машины необходимо преобразовать в данные чтения HiFi с помощью программы CCS в программном обеспечении SMRTlink на сервере или запустив отдельно установленное программное обеспечение CCS. Для платформ Pacbio Sequel IIe и Revio, поскольку сам инструмент секвенирования имеет встроенный вычислительный сервер, его можно настроить через SMRTlink перед запуском секвенирования, а данные считывания HiFi можно получить непосредственно после отключения машины.
Поэтому, когда вы получаете данные секвенирования PacBio, например, при загрузке общедоступных данных, особенно ранних данных, вы должны уточнить, является ли это дополнительным чтением или чтением HiFi. Данные, недавно полученные от поставщиков услуг секвенирования, обычно считываются HiFi после запуска программного обеспечения CCS.
Пользователи, у которых есть инструменты PacBio и их серверы настроены с помощью программного обеспечения SMRTlink, могут запустить программу CCS (циклического консенсусного секвенирования) непосредственно в SMRTlink. После завершения операции вы также получите отчет анализа CCS в SMRTlink, который предоставит информацию. HiFi читает информационно-статистическую информацию, отображение наглядных графиков.
Следующее руководство подготовлено нашими студентами и преподавателями, у которых нет инструмента секвенирования или установки и настройки программного обеспечения SMRTlink, но которые хотят установить и запустить программу CCS на своих собственных серверах или высокопроизводительных рабочих станциях.
4. Установка и использование программы CCS

Официальный сайт СКС:https://ccs.how/
Официальный сайт CCS (github):https://github.com/PacificBiosciences/ccs
1. Убедитесь, что миниконда установлена.
#Используйте conda напрямую, чтобы установить последнюю версию pbccs
$ conda install -c bioconda pbccs
#Версия 6.4.02. Работа программного обеспечения
Pacbio Sequel Данные о высадке II платформы представлены в формате bam. Файлы bam можно напрямую адаптировать к большинству последующих программ анализа. Файлы, в которых хранятся достоверные данные, обычно называются: *.subreads.bam, *.subreads.bam.pbi。
входной файл:sample.subreads.bam и соответствующий индексsample.subreads.bam.pbi
выходной файл:unaligned BAM (.bam);bgzipped FASTQ (.fastq.gz)。
Базовое использование, все параметры по умолчанию:
#генерировать .bam документ
$ ccs sample.subreads.bam sample.ccs.bam
#генерировать .fastq.gz документ
$ ccs sample.subreads.bam sample.hifi.fastq.gzРасширенное использование
#генерировать.bamдокумент
$ ccs --min-rq 0.99 --min-passes 3 -j 12 sample.subreads.bam sample.ccs.bam
#генерировать .fastq.gz документ
$ ccs --min-rq 0.999 --min-passes 5 -j 24 sample.subreads.bam sample.hifi.fastq.gz
#Следующие параметры часто устанавливаются и могут быть настроены в соответствии с потребностями данных и приложения. Остальные параметры могут быть установлены по умолчанию.
-j 12 Количество потоков ЦП
--min-passes 3 Минимальный создаваемый CCS read Количество дополнительных чтений, по умолчанию — 3.
--min-rq 0.99 Базовая точность, по умолчанию — 0,99, равная Q20.
--min-length Минимальная длина чтения, по умолчанию — 10.
--max-length Максимальная длина чтения, по умолчанию — 50000.CCS --help документ и параметры, при необходимости вы можете изменить их самостоятельно:
ccs - Generate circular consensus sequences (ccs) from subreads.
Usage:
ccs [options] <IN.subreads.bam|xml> <OUT.ccs.bam|fastq.gz|xml>
IN.subreads.bam|xml FILE Subreads (.subreads.bam or .subreadset.xml).
OUT.ccs.bam|fastq.gz|xml FILE Consensus reads (.bam, .fastq.gz, or .consensusreadset.xml).
Input Filter Options:
--min-passes INT Minimum number of full-length subreads required to generate CCS for a ZMW. [3]
--min-snr FLOAT Minimum SNR of subreads to use for generating CCS [2.5]
--top-passes INT Pick at maximum the top N passes for each ZMW. [60]
Draft Filter Options:
--min-length INT Minimum draft length before polishing. [10]
--max-length INT Maximum draft length before polishing. [50000]
Chunking Options:
--chunk STR Operate on a single chunk. Format i/N, where i in [1,N]. Examples: 3/24 or 9/9
--max-chunks Determine maximum number of chunks.
Model Override Options:
--model-path STR Path to a chemistry model file or directory containing model files.
--model-spec STR Name of chemistry or model to use, overriding default selection.
Processing Options:
--by-strand Generate a consensus for each strand.
--hd-finder Enable heteroduplex finder and splitting
--skip-polish Only output the initial draft template (faster, less accurate).
--all Emit all ZMWs.
--subread-fallback Emit a representative subread, instead of the draft consensus, if polishing failed.
--all-kinetics Calculate mean pulse widths (PW) and interpulse durations (IPD) for every ZMW.
--hifi-kinetics Calculate mean pulse widths (PW) and interpulse durations (IPD) for every HiFi read.
Output Filter Options:
--min-rq FLOAT Minimum predicted accuracy in [0, 1]. [0.99]
Output Files Options:
--report-file FILE Where to write the results report.
--report-json FILE Where to write the results report as json.
--metrics-json FILE Where to write the zmw metrics as json.
--suppress-reports Do not generate report or metric files per default, only those requested.
-h,--help Show this help and exit.
--version Show application version and exit.
-j,--num-threads INT Number of threads to use, 0 means autodetection. [0]
--log-level STR Set log level. Valid choices: (TRACE, DEBUG, INFO, WARN, FATAL). [WARN]
--log-file FILE Log to a file, instead of stderr.
Copyright (C) 2004-2022 Pacific Biosciences of California, Inc.
This program comes with ABSOLUTELY NO WARRANTY; it is intended for
Research Use Only and not for use in diagnostic procedures.5. Объяснение словаря на английском языке, связанного с режимом секвенирования CCS.
Что касается полимеразы, прочитайте, подчитайте оригинальное английское объяснение CCS.


Учебное пособие по Jetpack Compose для начинающих, базовые элементы управления и макет

Код js веб-страницы, фон частицы, код спецэффектов

【новый! Суперподробное】Полное руководство по свойствам компонентов Figma.
🎉Обязательно к прочтению новичкам: полное руководство по написанию мини-программ WeChat с использованием программного обеспечения Cursor.
[Забавный проект Docker] VoceChat — еще одно приложение для мгновенного чата (IM)! Может быть встроен в любую веб-страницу!

Как реализовать переход по странице в HTML (html переходит на указанную страницу)

Как решить проблему зависания и низкой скорости при установке зависимостей с помощью npm. Существуют ли доступные источники npm, которые могут решить эту проблему?

Серия From Zero to Fun: Uni-App WeChat Payment Practice WeChat авторизует вход в систему и украшает страницу заказа, создает интерфейс заказа и инициирует запрос заказа

Серия uni-app: uni.navigateЧтобы передать скачок значения

Апплет WeChat настраивает верхнюю панель навигации и адаптируется к различным моделям.

JS-время конвертации

Обеспечьте бесперебойную работу ChromeDriver 125: советы по решению проблемы chromedriver.exe не найдены

Поле комментария, щелчок мышью, специальные эффекты, js-код
Объект массива перемещения объекта JS

Как открыть разрешение на позиционирование апплета WeChat_Как использовать WeChat для определения местонахождения друзей
Я даю вам два набора из 18 простых в использовании фонов холста Power BI, так что вам больше не придется возиться с цветами!

Получить текущее время в js_Как динамически отображать дату и время в js

Вам необходимо изучить сочетания клавиш vsCode для форматирования и организации кода, чтобы вам больше не приходилось настраивать формат вручную.

У ChatGPT большое обновление. Всего за 45 минут пресс-конференция показывает, что OpenAI сделал еще один шаг вперед.
Copilot облачной разработки — упрощение разработки

Микросборка xChatGPT с низким кодом, создание апплета чат-бота с искусственным интеллектом за пять шагов

CUDA Out of Memory: идеальное решение проблемы нехватки памяти CUDA
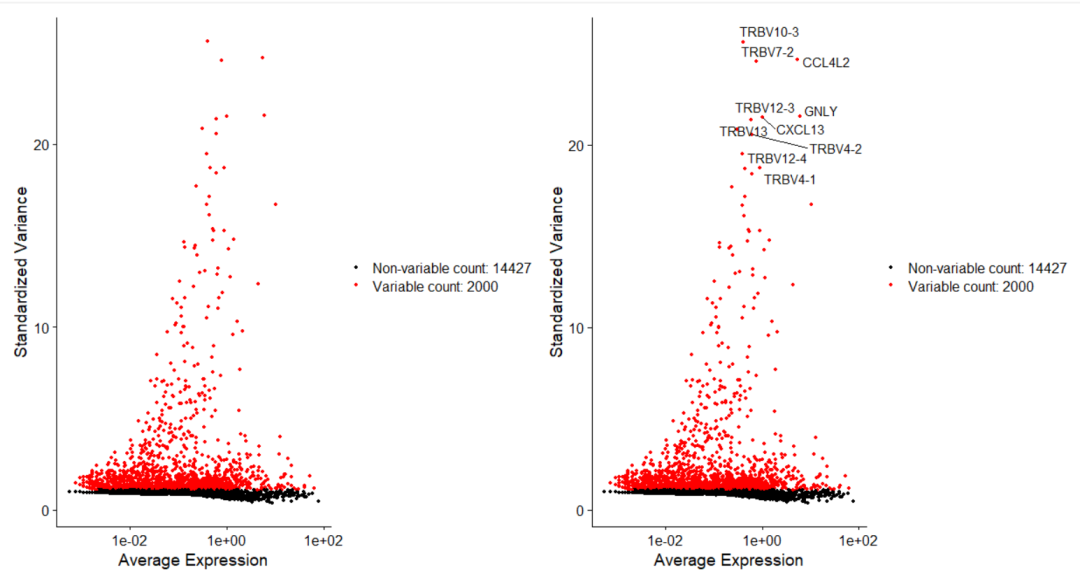
Анализ кластеризации отдельных ячеек, который должен освоить каждый&MarkerгенетическийВизуализация

vLLM: мощный инструмент для ускорения вывода ИИ

CodeGeeX: мощный инструмент генерации кода искусственного интеллекта, который можно использовать бесплатно в дополнение к второму пилоту.

Машинное обучение Реальный бой LightGBM + настройка параметров случайного поиска: точность 96,67%
Бесшовная интеграция, мгновенный интеллект [1]: платформа больших моделей Dify-LLM, интеграция без кодирования и встраивание в сторонние системы, более 42 тысяч звезд, чтобы стать свидетелями эксклюзивных интеллектуальных решений.
LM Studio для создания локальных больших моделей
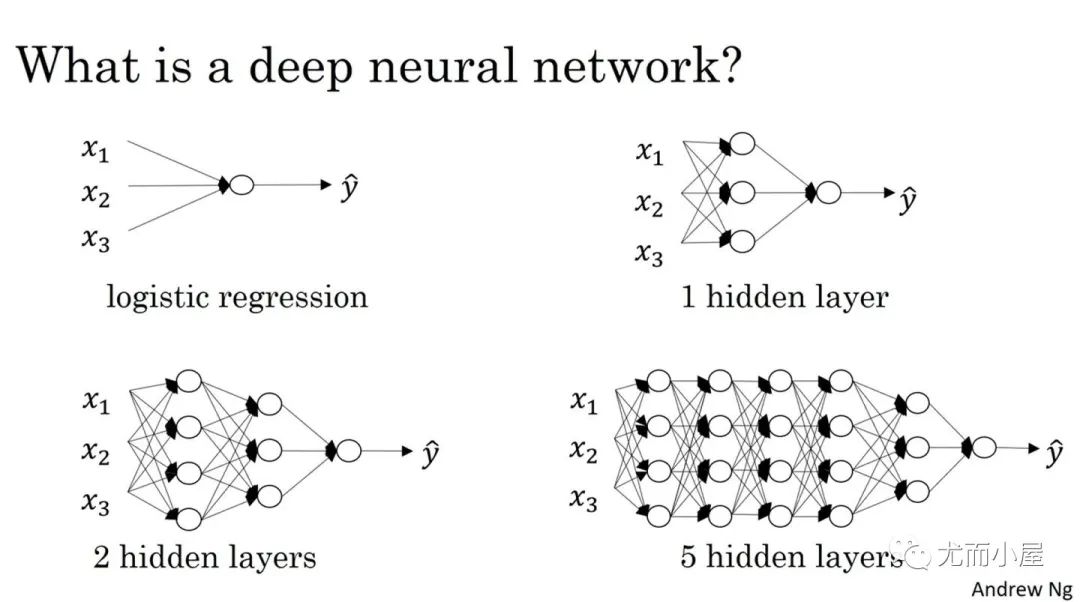
Как определить количество слоев и нейронов скрытых слоев нейронной сети?
