Полноразмерный транскриптом | Рабочий процесс анализа данных секвенирования третьего поколения Iso-Seq (PacBio) (1)
Я также считаю, что длинное секвенирование – это будущее секвенирования РНК! По мере снижения цен и улучшения качества основы традиционные методы секвенирования РНК второго поколения будут постепенно заменяться.
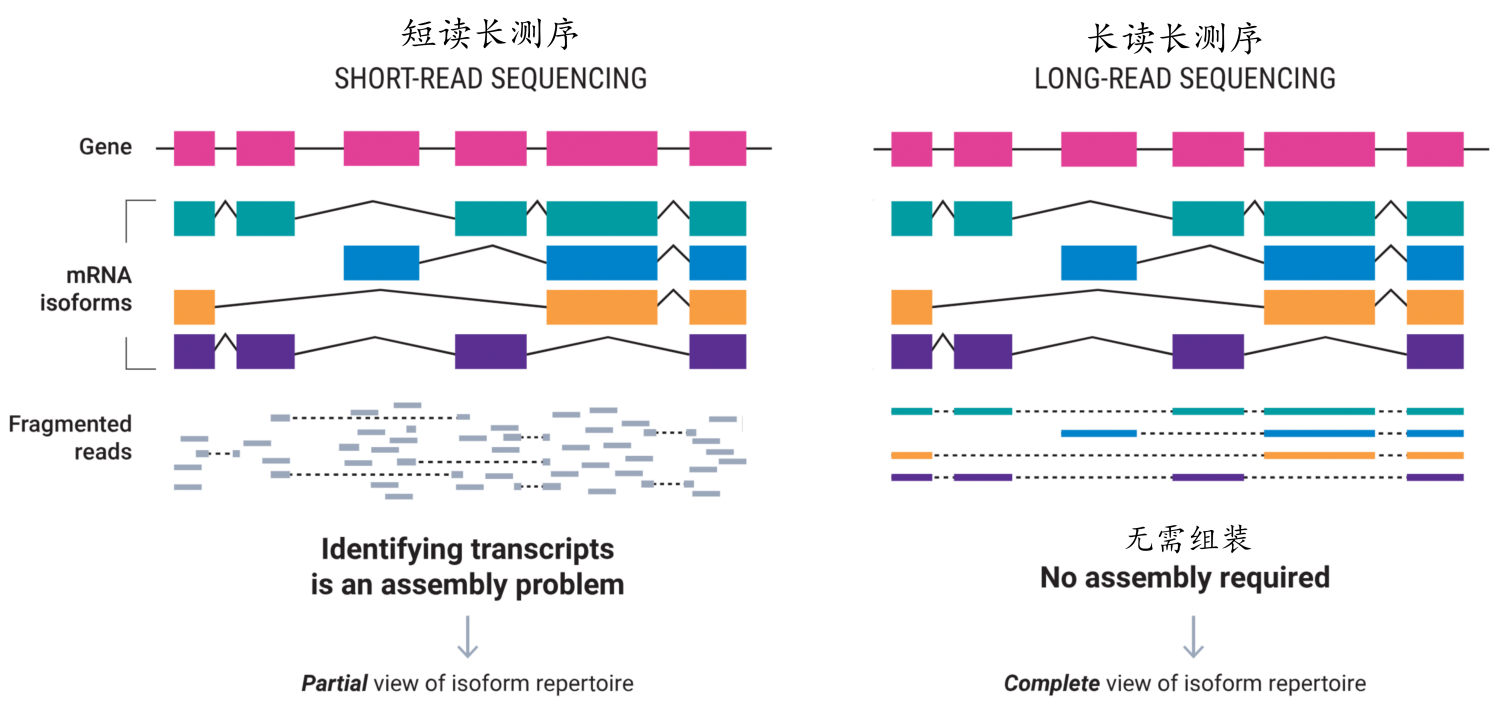
Транскрипты многих видов очень разнообразны и сложны. Подавляющее большинство эукариотических генов не соответствуют модели «один ген, один транскрипт». Эти гены часто имеют множественные формы альтернативного сплайсинга (AS). В настоящее время в различных исследованиях транскриптома широко используется технология секвенирования РНК (RNA-seq), основанная на технологии секвенирования второго поколения. Однако длина считывания последовательности при его секвенировании невелика (50-300 п.н.), и большинство из них могут охватывать лишь небольшую часть транскрипта, что затрудняет точную реконструкцию гомологичной изоформы (изоформы) того же транскрипта. Секвенирование РНК второго поколения делает реконструкцию полноразмерных транскриптов неточной и односторонней.
Полноразмерный транскриптом transcriptome)Секвенированиеианализироватьдана основеPacBioиOxford Nanoporeтри поколения Секвенированиеплатформа,Воспользуйтесь преимуществами длительного чтения,Нет необходимости прерывать РНК во время создания библиотеки и секвенирования.,Например, напрямую получите полноразмерную последовательность м РНК и полную структурную информацию, включая 5’UTR, 3’UTR и хвост поли А.,Это позволяет проводить точный анализ структурной информации, такой как гены альтернативного сплайсинга и слияния эталонных видов генома.,Преодолейте проблемы сборки коротких транскриптов и неполной информации для видов без эталонных геномов (Рисунок 1).
Первый PacBio О полном транскриптом Товар носит названиеIso-Seq, Полное имя Isoform-Sequencing, Это стандартизированное название технологии секвенирования транскриптов, разработанной собственными силами. Теперь используйте новейший комплект SMRTbell. prep kit 3.0руководить Секвенирование Строительство библиотеки。2023Год10Запущен в начале месяцаКонструктор библиотеки полноразмерной РНК Kinnex,Может объединить 5 транскриптов в одно чтение секвенирования.,В полной мере используйте преимущества длительного чтения.,Повышение производительности секвенирования,Используется с системой Revio.,Сделать количественную оценку полноразмерных транскриптов более практичной.
Метод Iso-Seq позволяет секвенировать целые молекулы к ДНК (до 10 т.п.н. и более) без необходимости сборки биоинформатических транскриптов, что позволяет секвенировать новые гены и изоформы в массовых и одноклеточных транскриптомах. Охарактеризовать организм и далее:
- Определение событий альтернативного сплайсинга (АС), включая переменные стартовые площадки、сайт завершения、События удержания интронов и пропуска экзонов.
- проходитьоткрытая рамка для чтения (ORF) Прогнозирование функционального воздействия новых изомеров.
- Обнаружение дифференциально экспрессируемых гомеоформ и событий переключения изоформ.
- Обнаружение событий слияния генов в образцах опухолей.
- Идентификация аллельных изомеров.
1. Экспериментальный процесс PacBio Iso-Seq
проходить PacBio SMRTbell prep kit 3.0 Создать библиотеку секвенирования Iso-Seq, подходящую для PacBio Sequel II и Revio Модель прибора (рис. 2).
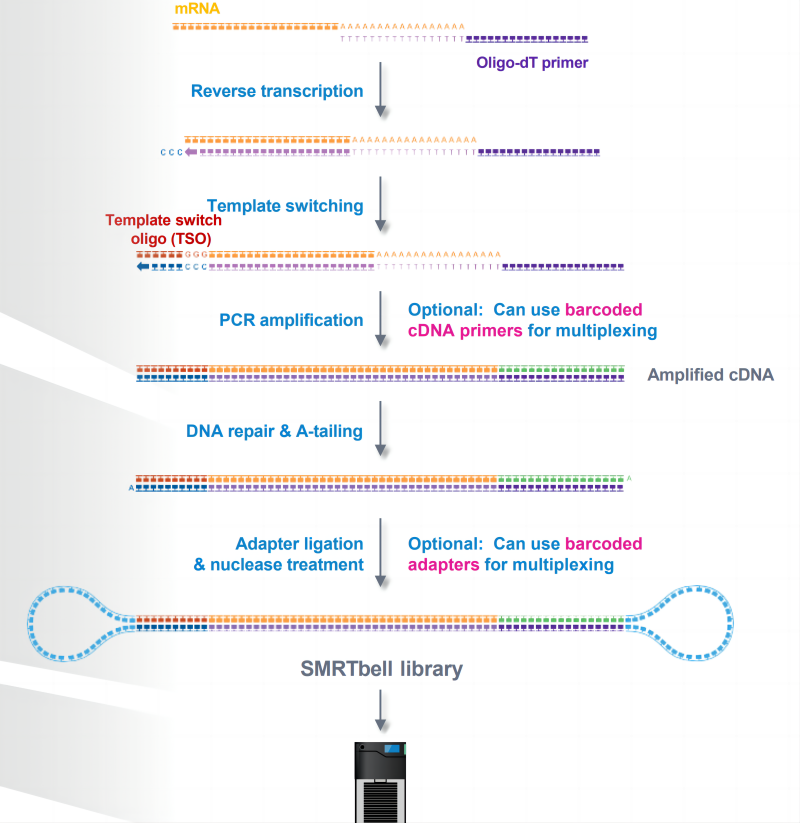
(1) использовать праймеры с обогащенным Oligo-dT для м РНК, содержащей хвост поли А.
(2) Используйте фермент Iso-Seq RT для обратной транскрипции м РНК.
(3) Добавьте олигопереключатель шаблона (Template Switch Oligo, TSO)
(4) провести ПЦР-амплификацию и обогащение синтезированной к ДНК,На этом этапе вы можете добавить штрих-код,Используется для смешивания образцов.
(5) Выполните восстановление повреждений, восстановление концов и А-хвост на полноразмерной к ДНК.
(6) Подключите адаптер для гантельного секвенирования SMRT и, наконец, объедините праймеры для секвенирования и свяжите ДНК-полимеразу, чтобы сформировать полную библиотеку секвенирования SMRT-bell.
2. Процесс анализа полноразмерного транскриптома Iso-Seq
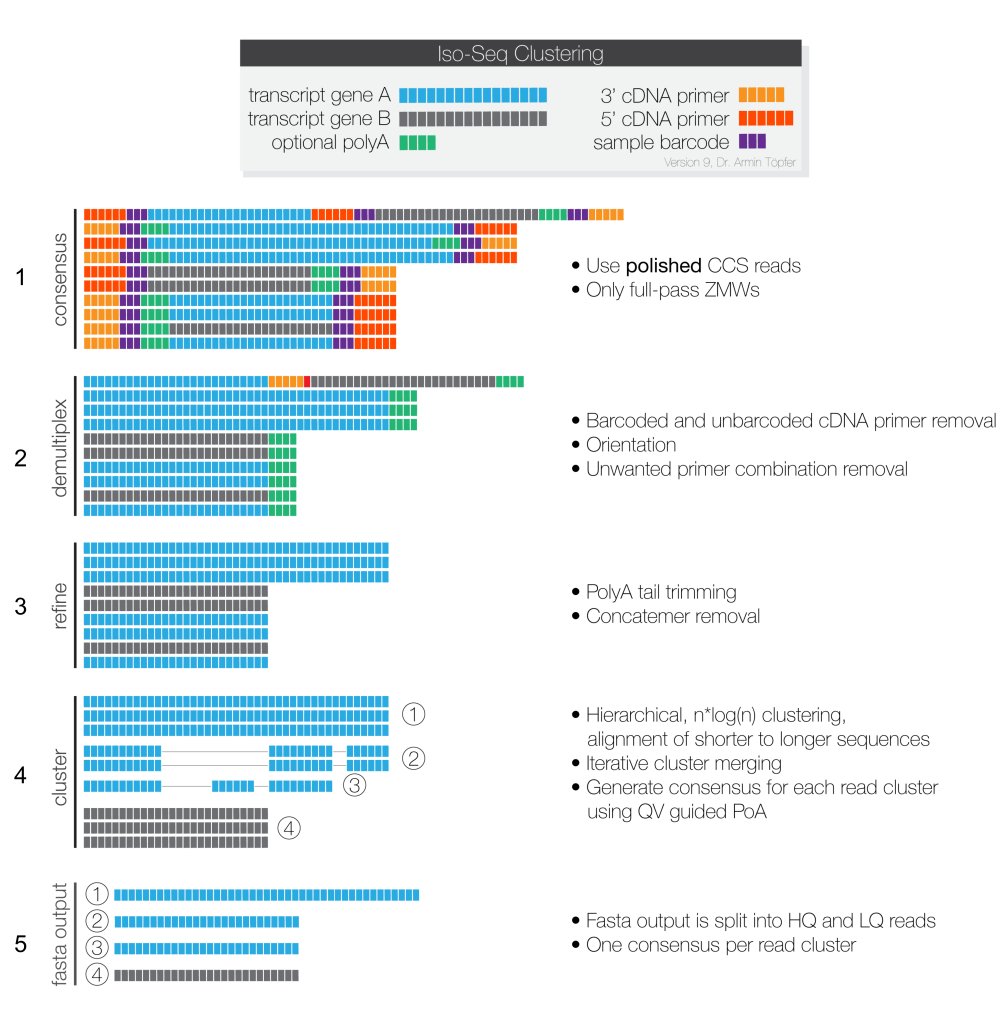
1. Основные понятия Iso-seq (1)
- ROI:reads of insert
ROI, полное название — чтения вставки, можно понимать как вставленные фрагменты. Сначала посмотрите на принципиальную схему считываний этапа построения библиотеки секвенирования третьего поколения, как показано на рисунке 3:
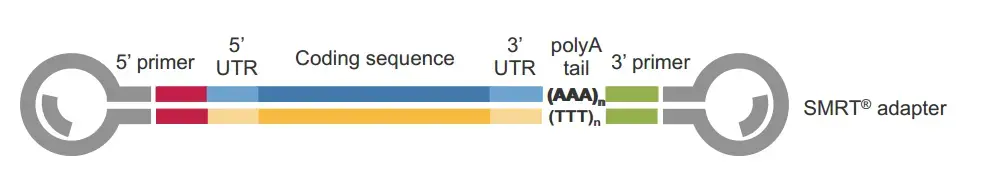
Для приведенных выше фрагментов библиотеки схематическая диаграмма ридов, генерируемых секвенированием, представлена на рисунке 4:
Поскольку это кольцевая молекула, циклическое секвенирование будет выполняться по ходу реакции секвенирования, если как положительная, так и отрицательная цепи вставленного фрагмента проверены один раз, будет выполнен полный проход; Для CCS для генерации чтений CCS требуется как минимум 2 полных прохода. Характерной чертой секвенирования третьего поколения является очень большая длина чтения, которая может достигать более десяти килобайт. Для коротких вставочных фрагментов проблем с определением CCS, конечно, нет, но для полноразмерных транскриптов - с длиной транскрипта. очень длинный, например, длина транскрипта 1 КБ, длина чтения 3 КБ. В настоящее время чтения, секвенированные в отверстии волновода с нулевой модой (ZMW), не могут достичь 2 полных проходов, и чтения CCS не могут быть сгенерированы. Чтобы решить эту проблему. и повысить коэффициент использования операций чтения, был предложен метод ROI Понятие ROI относится к вставленному фрагменту. ROI, полученная в результате секвенирования на рисунке 4, показана на рисунке 5:

ROI не требует двух полных проходов. По сравнению с CCS он больше подходит для анализа полноразмерных транскриптов.
- Артефакты, аномальные расшифровки, которые могут возникнуть во время строительства библиотеки.
Можно понять, что есть два источника:
Artificial Concatemer

Такая последовательность обусловлена этапом подготовки библиотеки, адаптера Последовательность неправильно связывает последовательности двух транскриптов с образованием кольцевой молекулы. Связано с концентрацией, обычно такое чтение Доля производимого продукта очень мала, менее 0,5%. При последующем анализе эта часть читается Необходимо удалить.
PCR Chimera

В реакции ПЦР, поскольку не полностью удлиненный продукт используется в качестве праймера для следующей реакции амплификации, появляется химерная последовательность. Интуитивно это означает, что продукт ПЦР происходит из двух или более прочтений. Химерная последовательность, генерируемая ПЦР, неизбежна в реакционной системе ПЦР и составляет около 3%. В последующем процессе анализа эту часть прочтений можно удалить с помощью программного обеспечения.
- FL Reads
FL, Полноценные чтения, полноценные транскрипты. От необработанных данных до рентабельности инвестиций, после удаления считываний артефактов из рентабельности инвестиций, мы получили чистые считывания для последующего анализа. Чистые чтения — это уже последовательности транскриптов. Давайте сначала посмотрим, какие чистые чтения являются полноразмерными транскриптами, а какие — нет.
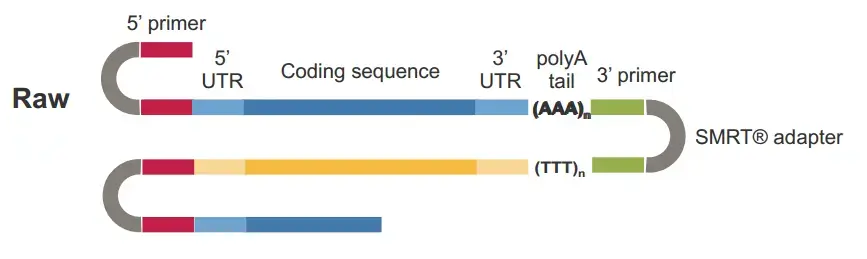
Схематическая диаграмма полноразмерного транскрипта, как показано на рисунке 8:

Для полноформатных стенограмм рентабельность инвестиций reads Содержит 5 футов primer и 3‘ primer; И появится поли А – структура (поли А; противmRNAичастьlncRNA)。
2. Программное обеспечение Iso-Seq.
Iso-SeqдаPacBioОфициально разработанныйPacBio subreads или HiFi Программное обеспечение, которое выполняет полноразмерный транскриптомный анализ данных и в конечном итоге выводит полноразмерные последовательности высококачественных транскриптов. По состоянию на 7 июня 2023 г. последняя версия — 4.0.0.
Домашняя страница Github:https://github.com/PacificBiosciences/IsoSeq
- Установка программного обеспечения: isoseqilima
#Используйте conda для установки isoseq, v4.0.0.
$ conda install -c bioconda isoseq
#lima, используется для дизассемблирования штрих-кода.
$ conda install -c bioconda lima- Процесс анализа Iso-Seq
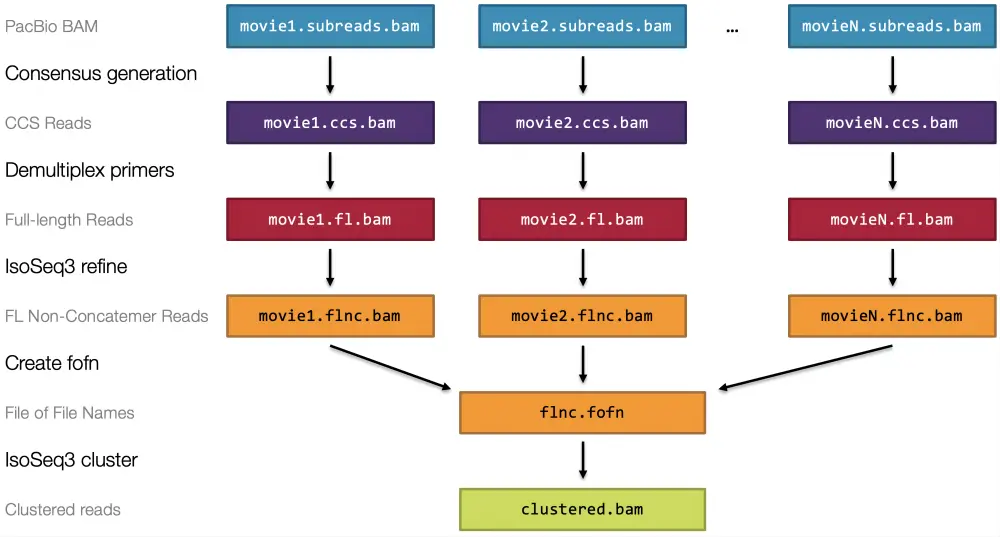
Весь процесс анализа Iso-Seq показан на рисунке 9:
(1) Исходные данные о высадке subreads.bam проходитьCCSприобретение программного обеспеченияHiFi Reads, hifi.reads.bam。
(2) Удалите праймеры на обоих концах к ДНК, разберите штрих-код и отрегулируйте направление транскрипта 5'-3' (поли А).
(3) удаление 3'-хвоста поли А и химерной (конкатемерной) последовательности.
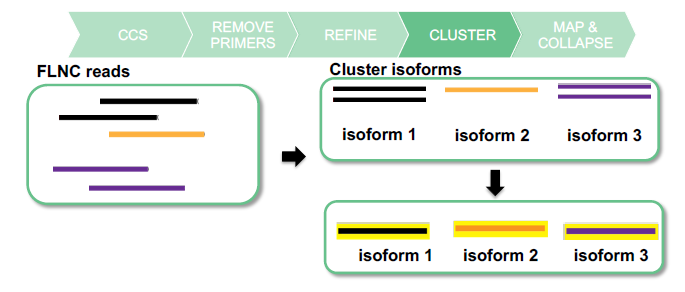
(4) Кластеризация транскриптов.
(5)ConsensusПоследовательность транскриптов.fastaформат вывода。
- Демонстрация данных официального образца одного образца
(1) Загрузка образца данных
# Download toy S-read dataset
# This is a toy dataset consisting of ~80k segmented reads (S-reads) from a Kinnex full-length RNA library
$ wget https://downloads.pacbcloud.com/public/dataset/IsoSeq_sandbox/human_80k_Sreads.segmented.bam
# Download the Iso-Seq v2 cDNA primers (from Iso-Seq express 2.0 kit)
$ wget https://downloads.pacbcloud.com/public/dataset/Kinnex-full-length-RNA/REF-primers/IsoSeq_v2_primers_12.fasta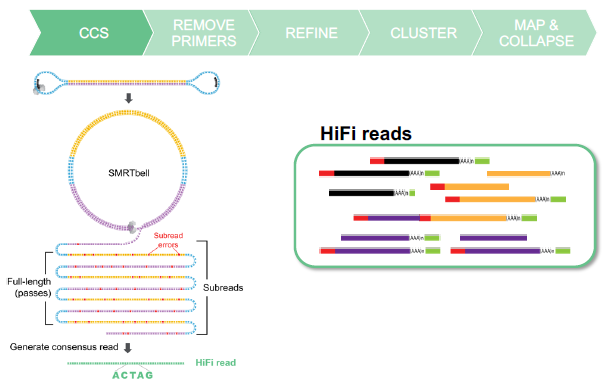
Hifi Оба конца чтения содержат 5' и 3' Праймерная последовательность.
(2) демультиплекс, используйте Лиму для разбора штрих-кода
$ lima --version
lima 2.9.0
$ lima human_80k_Sreads.segmented.bam IsoSeq_v2_primers_12.fasta human_80k.bam --isoseq --peek-guessДля данных iso-seq,использовать--isoseqдобавлять--peek-guessпараметры для снижения уровня ложноположительных результатов。Можетиспользовать --biosample-csv input.csvдобавлятьдобавлять Образец имени, bio sample name。
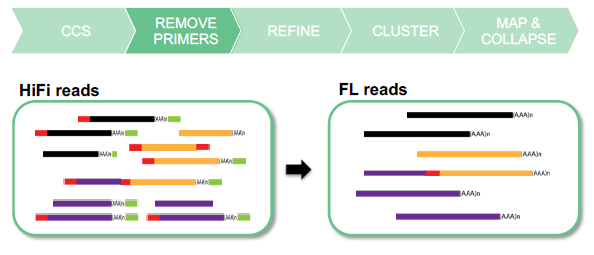
Demultplexи 5' - 3' После удаления праймера была получена хвостовая последовательность поли А. Full-Length reads (FL reads)。
(3)уточнить, использовать isoseq refineудалятьpoly(A)испаривающееся тело(concatemer)последовательность
- входной файлдля:
<primer--pair>.fl.bamиprimers.fasta。 - Выходные файлы в основном:
<movie>.flnc.bam。
$ isoseq refine human_80K.IsoSeqX_bc10_5p--IsoSeqX_3p.bam IsoSeq_v2_primers_12.fasta human_80K.flnc.bam --require-polya
$ ls human_80K.flnc.*
human_80K.flnc.bam
human_80K.flnc.bam.pbi
human_80K.flnc.consensusreadset.xml
human_80K.flnc.filter_summary.report.json
human_80K.flnc.report.csv--require-polya : Полными будут распознаваться только последовательности с хвостом поли А. длину и удалите последовательность поли А.
-j,--num-threads: Количество потоков ЦП.
--min-polya-length: Минимальная длина хвоста поли А, значение по умолчанию — 20.
3' приезжать после удаления хвоста поли А и химерной (конкатемерной) последовательности Full-Length Non-Concatemer (FLNC) reads。
(4) кластер, кластеризация транскриптов
- входной файл:
<movie>.flnc.bamorflnc.fofn。 - выходной файл:
<prefix>.bam
$ isoseq cluster2 human_80K.flnc.bam human_80K.transcripts.bam
$ ls human_80K.transcripts.*
human_80K.transcripts.bam
human_80K.transcripts.bam.pbi
human_80K.transcripts.cluster_report.csvИзоформы выходного транскрипта содержат не менее двух и более двух FLNC (полноразмерных неконкатемерная) поддержка последовательности. Если вы хотите включить синглтоны,Можетдобавлятьвходить--singletons。
Что касается того, какие условия выполняются, алгоритм кластеризации isoseq сгруппирует две последовательности в одну и ту же последовательность транскрипта (рис. 13):
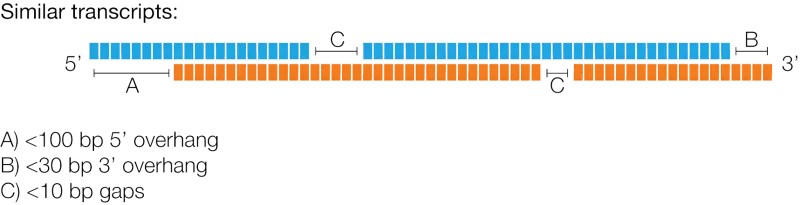
(А) Разница на 5'-концах составляет менее 100 п.н.
(B) Разница на 3'-концах составляет менее 30 п.н.
(C) Пробелы менее 10 б.п., верхнего предела количества пробелов нет.
- Демонстрация официального примера данных смешанной выборки
(1) Загрузка образца данных
# This is a 12-plex regular Iso-Seq (non-Kinex) run on Sequel II system consisting of ~3 million HiFi reads.
# Download HiFi reads from a non-Kinnex (regular Iso-Seq) BAM file
$ wget https://downloads.pacbcloud.com/public/dataset/Kinnex-full-length-RNA/DATA-SQ2-UHRR-Monomer/1-CCS/m64307e_230628_025302.hifi_reads.bam
$ wget https://downloads.pacbcloud.com/public/dataset/Kinnex-full-length-RNA/DATA-SQ2-UHRR-Monomer/1-CCS/m64307e_230628_025302.hifi_reads.bam.pbi
# Download the Iso-Seq v2 cDNA primers (from Iso-Seq express 2.0 kit)
$ wget https://downloads.pacbcloud.com/public/dataset/Kinnex-full-length-RNA/REF-primers/IsoSeq_v2_primers_12.fasta(2) демультиплекс, используйте Лиму для разбора штрих-кода
$ lima --version
lima 2.9.0
# Demux and primer removal
$ lima --isoseq --peek-guess m64307e_230628_025302.hifi_reads.bam IsoSeq_v2_primers_12.fasta UHRR.bamКаждыйиндивидуальныйbarcodeпара выходов одининдивидуальный.bamдокумент,общий12индивидуальный.bamФайловая переписка12индивидуальныйобразец。
(3) Имена файлов объединенных и разделенных образцов.
# Combine inputs
$ ls UHRR.IsoSeqX*bam > all.fofn
$ cat all.fofn
UHRR.IsoSeqX_bc01_5p--IsoSeqX_3p.bam
UHRR.IsoSeqX_bc02_5p--IsoSeqX_3p.bam
UHRR.IsoSeqX_bc03_5p--IsoSeqX_3p.bam
UHRR.IsoSeqX_bc04_5p--IsoSeqX_3p.bam
UHRR.IsoSeqX_bc05_5p--IsoSeqX_3p.bam
UHRR.IsoSeqX_bc06_5p--IsoSeqX_3p.bam
UHRR.IsoSeqX_bc07_5p--IsoSeqX_3p.bam
UHRR.IsoSeqX_bc08_5p--IsoSeqX_3p.bam
UHRR.IsoSeqX_bc09_5p--IsoSeqX_3p.bam
UHRR.IsoSeqX_bc10_5p--IsoSeqX_3p.bam
UHRR.IsoSeqX_bc11_5p--IsoSeqX_3p.bam
UHRR.IsoSeqX_bc12_5p--IsoSeqX_3p.bamfofn: сокращение от «файлы с именами файлов».
(4)уточнить, использовать isoseq refineудалятьpoly(A)испаривающееся тело(concatemer)последовательность
# Remove poly(A) tails and concatemer
$ isoseq refine all.fofn IsoSeq_v2_primers_12.fasta UHRR.flnc.bam --require-polya
$ ls UHRR.flnc.*
UHRR.flnc.bam
UHRR.flnc.bam.pbi
UHRR.flnc.consensusreadset.xml
UHRR.flnc.filter_summary.report.json
UHRR_80K.flnc.report.csv(5) кластер, кластеризация транскриптов
$ isoseq cluster2 UHRR.flnc.bam UHRR.transcripts.bam(6)Польский (по желанию)
$ isoseq cluster flnc.fofn clustered.bam --verbose --use-qvsздесьиспользоватьisoseq cluster,Вместодаisoseq cluster2, clusterпо сравнению сcluster2Больше времени уходит。PolishНемного улучшит качество данных,Нетда Необходимые шаги。 После завершения операции получаются следующие файлы:
<prefix>.bam<prefix>.hq.fasta.gzwith predicted accuracy ≥ 0.99<prefix>.lq.fasta.gzwith predicted accuracy < 0.99<prefix>.bam.pbi<prefix>.transcriptset.xml
Вкратце, как показано выше, обзор процесса анализа iso-seq и выходные файлы каждого этапа показаны на рисунке 16.
Ссылки
- Iso-seq Essential Foundation-blog.csdn
- Процесс анализа полноразмерных транскриптомных данных третьего поколения pacbio
- PacBio Iso-Seq Workshop Online


Учебное пособие по Jetpack Compose для начинающих, базовые элементы управления и макет

Код js веб-страницы, фон частицы, код спецэффектов

【новый! Суперподробное】Полное руководство по свойствам компонентов Figma.
🎉Обязательно к прочтению новичкам: полное руководство по написанию мини-программ WeChat с использованием программного обеспечения Cursor.
[Забавный проект Docker] VoceChat — еще одно приложение для мгновенного чата (IM)! Может быть встроен в любую веб-страницу!

Как реализовать переход по странице в HTML (html переходит на указанную страницу)

Как решить проблему зависания и низкой скорости при установке зависимостей с помощью npm. Существуют ли доступные источники npm, которые могут решить эту проблему?

Серия From Zero to Fun: Uni-App WeChat Payment Practice WeChat авторизует вход в систему и украшает страницу заказа, создает интерфейс заказа и инициирует запрос заказа

Серия uni-app: uni.navigateЧтобы передать скачок значения

Апплет WeChat настраивает верхнюю панель навигации и адаптируется к различным моделям.

JS-время конвертации

Обеспечьте бесперебойную работу ChromeDriver 125: советы по решению проблемы chromedriver.exe не найдены

Поле комментария, щелчок мышью, специальные эффекты, js-код
Объект массива перемещения объекта JS

Как открыть разрешение на позиционирование апплета WeChat_Как использовать WeChat для определения местонахождения друзей
Я даю вам два набора из 18 простых в использовании фонов холста Power BI, так что вам больше не придется возиться с цветами!

Получить текущее время в js_Как динамически отображать дату и время в js

Вам необходимо изучить сочетания клавиш vsCode для форматирования и организации кода, чтобы вам больше не приходилось настраивать формат вручную.

У ChatGPT большое обновление. Всего за 45 минут пресс-конференция показывает, что OpenAI сделал еще один шаг вперед.
Copilot облачной разработки — упрощение разработки

Микросборка xChatGPT с низким кодом, создание апплета чат-бота с искусственным интеллектом за пять шагов

CUDA Out of Memory: идеальное решение проблемы нехватки памяти CUDA
Анализ кластеризации отдельных ячеек, который должен освоить каждый&MarkerгенетическийВизуализация

vLLM: мощный инструмент для ускорения вывода ИИ

CodeGeeX: мощный инструмент генерации кода искусственного интеллекта, который можно использовать бесплатно в дополнение к второму пилоту.

Машинное обучение Реальный бой LightGBM + настройка параметров случайного поиска: точность 96,67%
Бесшовная интеграция, мгновенный интеллект [1]: платформа больших моделей Dify-LLM, интеграция без кодирования и встраивание в сторонние системы, более 42 тысяч звезд, чтобы стать свидетелями эксклюзивных интеллектуальных решений.
LM Studio для создания локальных больших моделей
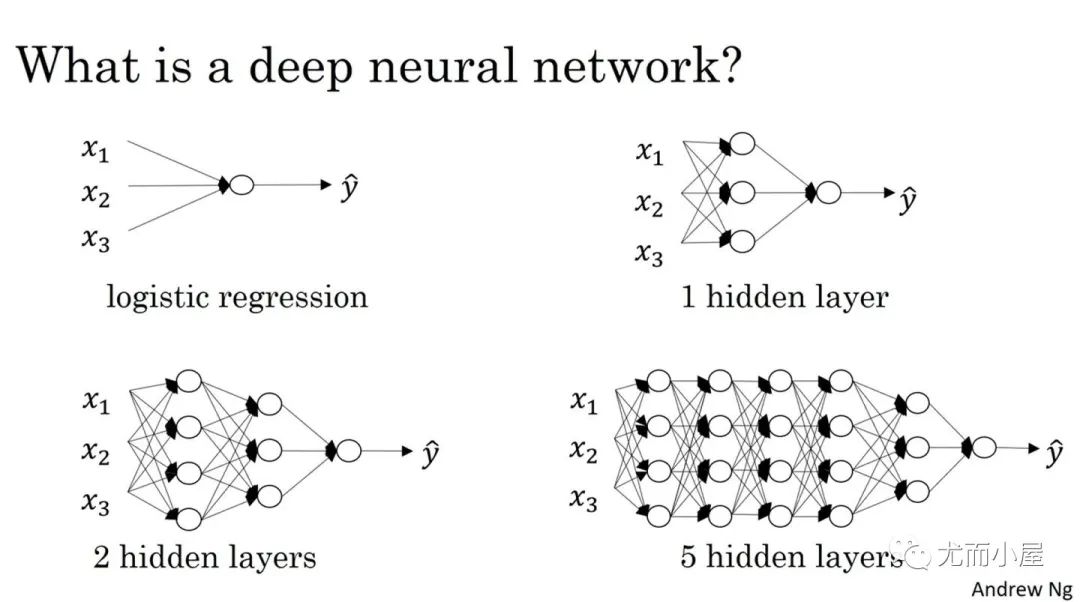
Как определить количество слоев и нейронов скрытых слоев нейронной сети?
