Полноразмерный транскриптом | Процесс анализа полноразмерного транскриптома третьего поколения Oxford Nanopore (ONT) — контроль качества и предварительная обработка данных
Полноразмерное секвенирование транскриптома ONT относится к полноразмерному секвенированию транскриптома на основе платформы секвенирования третьего поколения Oxford Nanopore Technologies (ONT). Благодаря использованию долгочитаемых характеристик платформы секвенирования третьего поколения нет необходимости фрагментировать транскрипты и получить высококачественную полноразмерную последовательность транскриптома от 5'-конца до 3'-конца м РНК определенного вида ( или дн РНК с поли А-хвостом) можно получить напрямую. Информация (рис. 1), которая может точно идентифицировать изменения в структуре транскрипта, такие как альтернативный сплайсинг, слияние генов, семейство генов, альтернативное полиаденилирование (APA), аллель-специфическая экспрессия и т. д. Полноразмерное секвенирование транскриптома на основе платформы секвенирования третьего поколения ONT позволяет не только точно идентифицировать вышеупомянутые структурные вариации транскриптов, но также достичь уровней экспрессии транскриптов (м РНК или поли А+ дн РНК) благодаря текущей стоимости секвенирования и пропускной способности (относительно платформа PacBio). Точная количественная оценка и дифференциальный анализ.
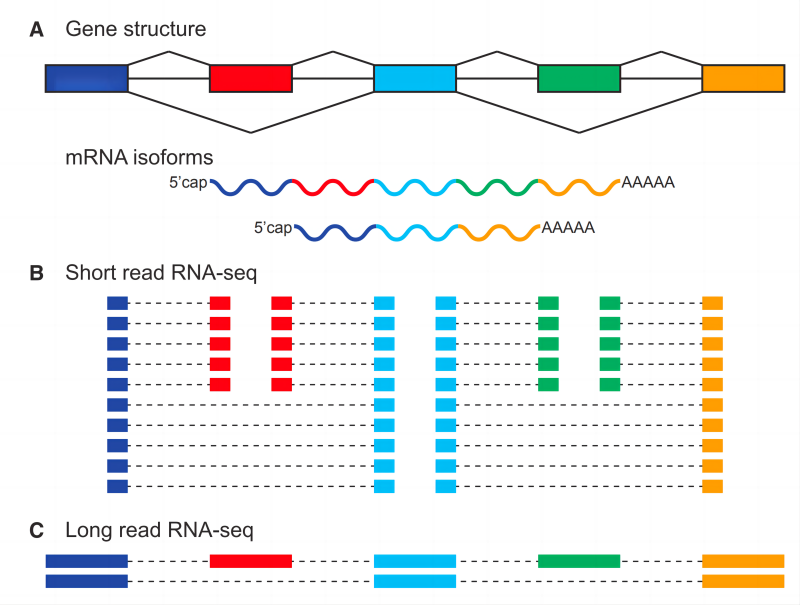
1. Введение в технологию секвенирования ONT.
Секвенирование ONT — это технология секвенирования третьего поколения, основанная на распознавании электрических сигналов базовых последовательностей. Существуют различия в химических свойствах различных оснований ДНК/РНК. Под руководством молекулярных двигателей одна молекула нуклеиновой кислоты связывается и раскручивает белок нанопоры, встроенный в биологическую мембрану. При прохождении через канал нанопоры возникают препятствия. основания изменяются, образуя характерный сигнал изменения ионного тока. Обнаружив эти сигналы в режиме реального времени, можно получить соответствующие базовые типы и завершить секвенирование (рис. 2). В настоящее время базы считываются с помощью сложного алгоритма под названием «Рекуррентная нейронная сеть».
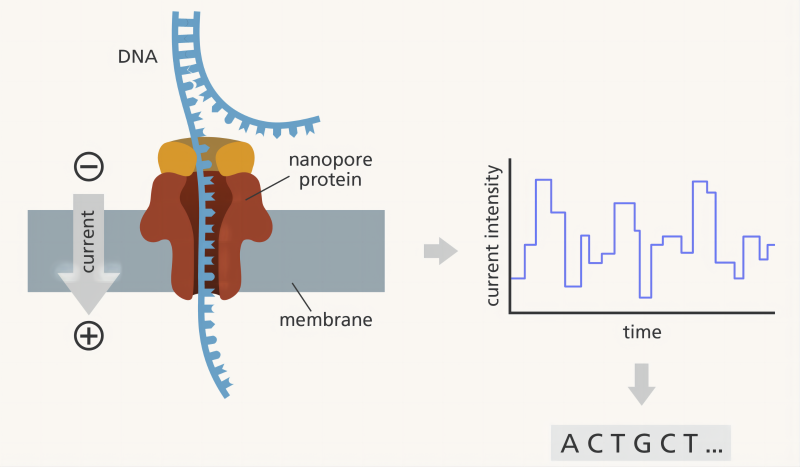
Технология секвенирования ONT имеет следующие характеристики:: 1) Длинная длина чтения, самая длинная длина чтения может достигать 4,2. Уровни выше M способствуют обнаружению структурных вариаций, таких как альтернативный сплайсинг и слияние генов; 2) Низкая стоимость: по сравнению с другими технологиями секвенирования третьего поколения обработка образцов для секвенирования ONT чрезвычайно проста, без необходимости использования ДНК-полимеразы, лигазы и dNTP, а цена секвенирования низкая; 3) ПЦР-амплификация не требуется во избежание ошибок или изменений численности, которые могут быть вызваны ПЦР-амплификацией при секвенировании второго поколения; 4) прямое построение библиотеки РНК/ДНК позволяет напрямую считывать информацию о модификациях оснований, например, о модификациях метилирования 5mC, 6mA и т. д., без необходимости проведения экспериментов по конверсии бисульфата или иммунопреципитации, таких как секвенирование второго поколения; 5) Отсутствует содержание GC и предпочтение оснований, а количественная оценка экспрессии транскрипта является точной.
2. Экспериментальный процесс полноразмерного секвенирования транскриптома ONT.
Следуйте документу по созданию библиотеки к ДНК-ПЦР, предоставленному Oxford Nanopore Technologies (ONT) (последняя версия — SQK-PCS114), включая тестирование качества образцов, создание библиотеки, тестирование качества библиотеки, секвенирование библиотеки и т. д., а также отображение в режиме к ДНК-ПЦР. (Рисунок 3). Начните с 10 нг обогащенной РНК (обогащенной поли А или обедненной рибосомами) или 500 нг общей РНК.
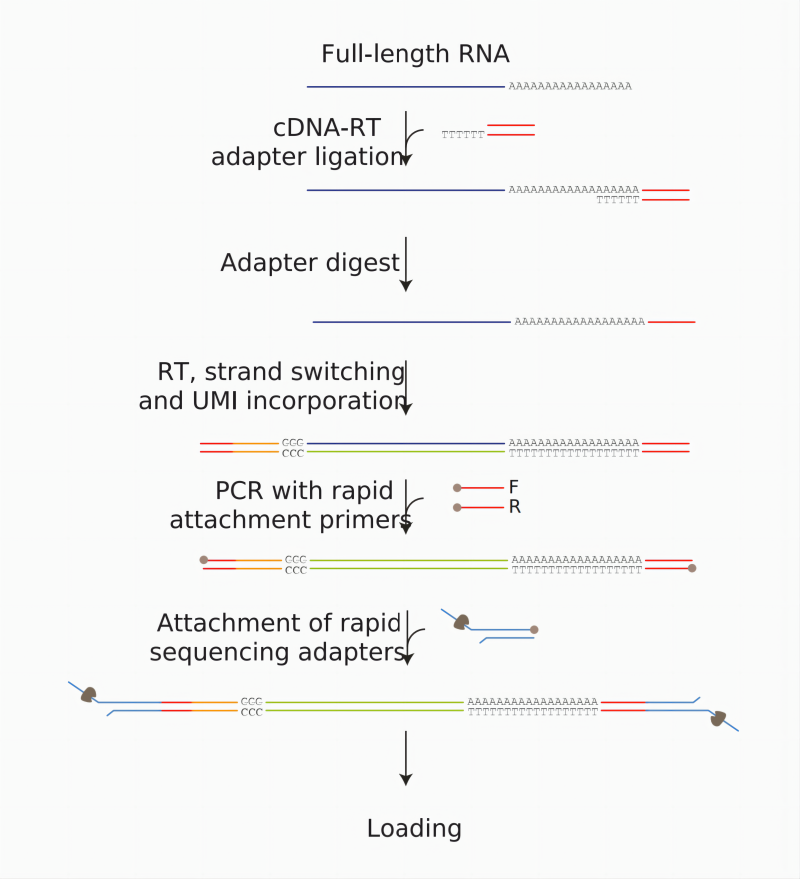
3. Процесс анализа полноразмерного транскриптома ONT
PacBioполный транскриптом официально разработал собственное оптимизированное программное обеспечение и процесс кластеризации транскриптов.,IsoSeq(https://isoseq.how/)。ONTполный Анализ транскриптом больше полагается на программное обеспечение и процессы, разработанные третьими сторонами.,Он основан наminimap2иStringTie2построен wf-transcriptomesпроцесс Развернуто вepi2me-labsдля пользователей。
Существующее программное обеспечение для восстановления и реконструкции полноразмерных транскриптов можно условно разделить на две категории. Один тип опирается на эталонный геном и файлы его аннотаций.,нравитьсяFLAIR,TAMA,TALONждать;Одна категория – этоreference-free,нравитьсяIsoSeq, LyRic。Есть и старые бренды.RNA-seqПрограммное обеспечение для анализа,StringTie2,Можно комбинироватьshort считывает (данные секвенирования второго поколения) и long reads(секвенирование третьего поколенияданные);IsoQuant и StringTie2Возможность полагаться на аннотации эталонного генома.,Также Ноde novo реконструкция полноразмерных транскриптов без аннотаций.
ONTполный транскриптомданныеДо анализа,Необходимо провести контроль качества офлайн-данных и идентифицировать полноразмерные транскрипты.,может использоваться в качестве входного файла для вышеуказанного программного обеспечения.,На этот раз мы сначала выполняем контроль качества и предварительную обработку данных ONT.,Подготовьте свои данные,Провести дальнейший анализ。потому чтоIsoQuant,Bambu,FLAIR,TAMA,TALONждать Программное обеспечение подходит для обоихONTТакже относится кPacBioизполный транскриптомданных, поэтому позже я дам руководство по использованию каждого программного обеспечения.
Демонстрационные данные полноразмерного транскриптома ONT предоставлены Euroopen. Nucleotide Archive(ENA)данные Библиотека:PRJEB31662,СкачаноERR3218377.fastq.gz,ERR3218373.fastq.gzдва образцаизданныев качестве демонстрации。
1. Исходные офлайн-данные fast5 -- Dorado
нравиться Если это оригинализfast5данные,Нужно использоватьDorado(илиGUPPY)Программное обеспечение преобразует электрические сигналы в базовые последовательности.。Вообще говоря,Поставщики услуг секвенирования разделятbarcodeиз.bam или/и .fastq Файл предоставляется пользователю, и пользователю не нужно его конвертировать самостоятельно.
2. Оригинальная статистика качества офлайн-данных — NanoComp.
О статистике качества и визуализации данных об отключениях ONT,Пожалуйста, обратитесь к предыдущемуиз Подробное руководство:секвенирование третьего поколения - Oxford Nanopore (ONT) анализ данных - данные Контроль качестваифильтр。Используйте его прямо здесьNanoCompОдин шаг:
- Используйте программное обеспечение NanoComp:https://github.com/wdecoster/nanocomp
- Установка программного обеспечения
$ pip install NanoComp- Использование программного обеспечения
#Официальный пример использования
$ NanoComp --bam alignment1.bam alignment2.bam alignment3.bam --outdir compare-runs
$ NanoComp --fastq reads1.fastq.gz reads2.fastq.gz reads3.fastq.gz reads4.fastq.gz --names run1 run2 run3 run4
#актуальный образец
$ nohup NanoComp -t 24 -f pdf \
--fastq 0_raw_fq/ERR3218373.fastq.gz 0_raw_fq/ERR3218377.fastq.gz \
--names ERR3218373 ERR3218377 \
-o NanoComp &
# -f Изображение выводится в формате PDF: {'png' (по умолчанию), 'jpg', 'jpeg', 'webp', 'svg', 'pdf', 'eps', 'json'}
# -t Количество запущенных потоков
# -o, --outdir OUTDIR Папка с выходным файломЕсли качество данных секвенирования и длина чтения соответствуют ожидаемым стандартам, перейдите к следующему шагу.
3. Контроль качества исходных офлайн-данных -- Chopper
Фильтровать исходную последовательность высадки (по длине или/и среднее качество базы),резать(Последовательность головы и хвоста)ипоследовательность загрязнения(--contam)из Удалить можно использоватьChopper。
- Используйте программное обеспечение chopper:https://github.com/wdecoster/chopper
- Установка программного обеспечения
$ conda install -c bioconda chopper- Использование программного обеспечения
#Официальный пример использования
$ gunzip -c reads.fastq.gz | chopper -q 10 -l 500 | gzip > filtered_reads.fastq.gz
#актуальный образец
$ gunzip -c 0_raw_fq/ERR3218373.fastq.gz | chopper -q 7 -l 50 | gzip > 1_chopper/ERR3218373.fastq.gz &
$ gunzip -c 0_raw_fq/ERR3218377.fastq.gz | chopper -q 7 -l 50 | gzip > 1_chopper/ERR3218377.fastq.gz &
#Результаты запуска
Kept 291095 reads out of 525588 reads
Kept 267866 reads out of 604601 reads- Удалить операции чтения, среднее базовое качество которых меньше 7. Обычно это значение является значением по умолчанию для автономных данных, поэтому, если вы хотите выполнить более строгий контроль качества, вы можете установить это значение больше.
- Удалить последовательности с длиной чтения менее 50 пар оснований.,Это значение также можно настроить самостоятельно. Удалите чтение небольших фрагментов и чтение с высокой степенью ухудшения качества.,Уменьшите неспецифические выравнивания.
- нравиться Если присутствует во время секвенированияпоследовательность загрязнения Можетпроходить
--contamудалить определенныепоследовательность загрязнения,Например, последовательности заражения микоплазмой.
4. Идентификация полноразмерной последовательности транскрипта - Pychopper
PychopperЭто идентификация,Ориентацияи Обрезать по всей длинеNanopore Инструмент для последовательностей к ДНК, который также может восстанавливать слитые последовательности.
- Используйте программное обеспечение
Pychopper:https://github.com/epi2me-labs/pychopper
- Установка программного обеспечения
$ conda install -c nanoporetech -c conda-forge -c bioconda "nanoporetech::pychopper"- Использование программного обеспечения
#Официальный пример использования
#Example usage with default PCS109/DCS109 primers using the default pHMM backend.
$ pychopper -r report.pdf -u unclassified.fq -w rescued.fq input.fq full_length_output.fq
#актуальный образец
$ mkdir 2_pychopper
$ nohup pychopper -t 8 -r 2_pychopper/ERR3218373_report.pdf \
-u 2_pychopper/ERR3218373_unclassified.fq \
-w 2_pychopper/ERR3218373_rescued.fq \
1_chopper/ERR3218373.fastq.gz 2_pychopper/ERR3218373_full_length.fq \
1> 2_pychopper/ERR3218373.pychopper.log 2>&1 &
$ nohup pychopper -t 8 -r 2_pychopper/ERR3218377_report.pdf \
-u 2_pychopper/ERR3218377_unclassified.fq \
-w 2_pychopper/ERR3218377_rescued.fq \
1_chopper/ERR3218377.fastq.gz 2_pychopper/ERR3218377_full_length.fq \
1> 2_pychopper/ERR3218377.pychopper.log 2>&1 &
# -r report_pdf Report PDF (pychopper_report.pdf).
# -u unclass_output Write unclassified reads to this file
# -w rescue_output Write rescued reads to this file.
# -Q min_qual Minimum mean base quality (7.0).
# -z min_len Minimum segment length (50).pychopperОбнаруживается только двустороннийиз Идентификация праймера определяется как полноразмерная полная.(full length)изпоследовательность,Поэтому не обрезайте и не удаляйте праймеры на обоих концах последовательности, прежде чем делать это.,Включено в использованиеDoradoЭлектрический сигнал преобразует основанияизкогда。- проходить
-Qи-zможет быть достигнутоchopperиз Функция,Поэтому, если нет удаления загрязняющей последовательности,Вы можете пропустить вторую часть и использовать ее напрямую.pychopper。 - -k может указать комплект и протокол. комплект{PCS109, PCS110, PCS111, LSK114}.
- Окончательная полноразмерная последовательность транскрипта
full_lenght.fastqможно использовать для последующего анализа.
Ссылки:
- Полноразмерный транскриптом третьего поколения нанопор
- Серия 1 полноразмерного секвенирования транскриптома ONT: Первое введение
- Новый инструмент для прогнозирования структуры генов — полноразмерный транскриптом ONT
- Park, Eddie et al. "The expanding landscape of alternative splicing variation in human populations." The American Journal of Human Genetics (2018).
- Mestre-Tomás, Jorge, et al. "SQANTI-SIM: a simulator of controlled transcript novelty for lrRNA-seq benchmark." Genome Biology 24.1 (2023): 286.
- Полноразмерное секвенирование транскриптома нанопор


Учебное пособие по Jetpack Compose для начинающих, базовые элементы управления и макет

Код js веб-страницы, фон частицы, код спецэффектов

【новый! Суперподробное】Полное руководство по свойствам компонентов Figma.
🎉Обязательно к прочтению новичкам: полное руководство по написанию мини-программ WeChat с использованием программного обеспечения Cursor.
[Забавный проект Docker] VoceChat — еще одно приложение для мгновенного чата (IM)! Может быть встроен в любую веб-страницу!

Как реализовать переход по странице в HTML (html переходит на указанную страницу)

Как решить проблему зависания и низкой скорости при установке зависимостей с помощью npm. Существуют ли доступные источники npm, которые могут решить эту проблему?

Серия From Zero to Fun: Uni-App WeChat Payment Practice WeChat авторизует вход в систему и украшает страницу заказа, создает интерфейс заказа и инициирует запрос заказа

Серия uni-app: uni.navigateЧтобы передать скачок значения

Апплет WeChat настраивает верхнюю панель навигации и адаптируется к различным моделям.

JS-время конвертации

Обеспечьте бесперебойную работу ChromeDriver 125: советы по решению проблемы chromedriver.exe не найдены

Поле комментария, щелчок мышью, специальные эффекты, js-код
Объект массива перемещения объекта JS

Как открыть разрешение на позиционирование апплета WeChat_Как использовать WeChat для определения местонахождения друзей
Я даю вам два набора из 18 простых в использовании фонов холста Power BI, так что вам больше не придется возиться с цветами!

Получить текущее время в js_Как динамически отображать дату и время в js

Вам необходимо изучить сочетания клавиш vsCode для форматирования и организации кода, чтобы вам больше не приходилось настраивать формат вручную.

У ChatGPT большое обновление. Всего за 45 минут пресс-конференция показывает, что OpenAI сделал еще один шаг вперед.
Copilot облачной разработки — упрощение разработки

Микросборка xChatGPT с низким кодом, создание апплета чат-бота с искусственным интеллектом за пять шагов

CUDA Out of Memory: идеальное решение проблемы нехватки памяти CUDA
Анализ кластеризации отдельных ячеек, который должен освоить каждый&MarkerгенетическийВизуализация

vLLM: мощный инструмент для ускорения вывода ИИ

CodeGeeX: мощный инструмент генерации кода искусственного интеллекта, который можно использовать бесплатно в дополнение к второму пилоту.

Машинное обучение Реальный бой LightGBM + настройка параметров случайного поиска: точность 96,67%
Бесшовная интеграция, мгновенный интеллект [1]: платформа больших моделей Dify-LLM, интеграция без кодирования и встраивание в сторонние системы, более 42 тысяч звезд, чтобы стать свидетелями эксклюзивных интеллектуальных решений.
LM Studio для создания локальных больших моделей
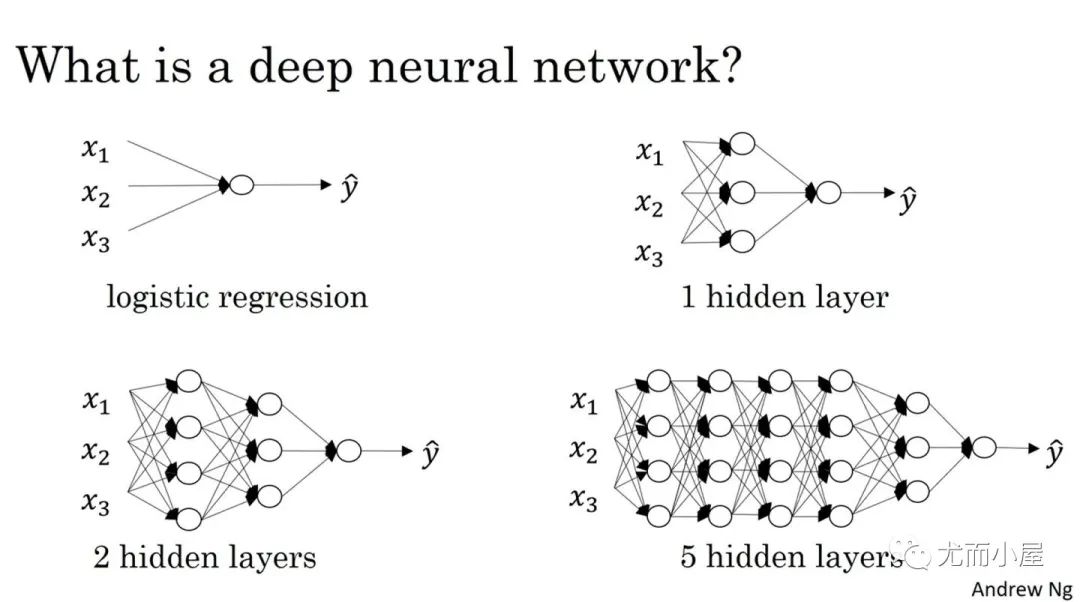
Как определить количество слоев и нейронов скрытых слоев нейронной сети?
