Полноразмерный транскриптом | Принципы технологии прямого секвенирования РНК (DRS), анализ данных и приложения ONT
«Платформа секвенирования третьего поколения, разработанная Oxford Nanopore Technologies (ONT), в настоящее время является единственной технологической платформой, которая может напрямую секвенировать природные цепи РНК. ONT - технология прямого секвенирования РНК (DRS, прямое секвенирование РНК) может секвенировать естественную полноразмерную РНК. Цепь может одновременно сохранять и обнаруживать информацию о модификации оснований РНК и может относительно точно оценивать длину поли(А)-хвоста, восстанавливая тем самым истинные характеристики РНК».

существуют в течение последнего десятилетия,RNAСеквенирование(
RNA-seq)постепенностановиться Анализировать дифференциальную экспрессию генов на уровне всего транскриптома.и ИсследоватьmRNAдифференциальное сращиваниеиз Нетилинедостатокизинструмент。Благодаря второму поколению высокопроизводительных Секвенированиетехнология(также известный какNext-Generation Sequencing,НГС) из Развитиястановитьсякнигаизредуцировать,Области применения секвенирования РНК также постоянно расширяются. Сейчас существуют,RNA-seq применялся во многих исследованиях на уровне РНК.,Включает экспрессию генов в одной клетке(single cell)、RNAкоманда переводчиков(translatome)и RNAструктура Группа(structurome)ждать。Рост в последние годыиз(захватывающийиз)Также новые приложения ВоляRNA-seqВ трехмерное пространствомежду,нравитьсянулевоймеждуизменятьзаписывать Группаизучать(spatialomics)。 Объединяя всё больше и большестановиться СпелыйизСеквенирование длинного чтения третьего поколения (long-read)и Прямое секвенирование РНК (Прямое RNA-seq)технология(картина1),и более продвинутые инструменты расчета и анализа.,RNA-seqВоля Помогите исследователямRNAБиология имеет более всеобъемлющее、более изысканныйизпонимать: отстенограммасуществовать, когда и где переписаноприезжатьсворачивание РНКа такжеМолекулярные взаимодействия выполняют функцииждать(1-2)。
в последние годы,С развитием технологии Секвенирования второго поколения,Традиционный транскриптомный анализ (RNA-seq) стал основным техническим средством изучения регуляции экспрессии генов. Регуляция генов очень разнообразна и сложна у многих видов.,Подавляющее большинство эукариотических генов не соответствуют модели «один ген — один ген».,Эти гены часто существуют в множественных сплайсированных формах. Автор: Секвенирование,Может очень точно проводить экспрессию генов и количественные исследования.,Но из-за Секвенирования длина чтения превышена,Невозможно точно получить полную информацию о стенограмме.,Потому что невозможно провести исследование на уровне стенограммы (рис. 1). поэтому,на основеПлатформа для секвенирования длинного считывания третьего поколенияизполный транскриптомстановитьсядля новогоиз Исследоватьподъем。
полный транскриптом(Full-length транскриптом) основан на PacBio(Pacific Biosciences) или ONT(Oxford Nanopore Technologies) Платформа секвенирования третьего поколения может напрямую получать полноразмерную последовательность и полную структурную информацию м РНК, включая 5'UTR, 3'UTR и хвост поли А, не прерывая сплайсинг после обогащения м РНК, тем самым точно анализируя альтернативные гены сплайсинга и слияния Структурная информация позволяет преодолеть проблемы сплайсинга коротких транскриптов и неполной информации для видов без эталонных геномов (рис. 2).
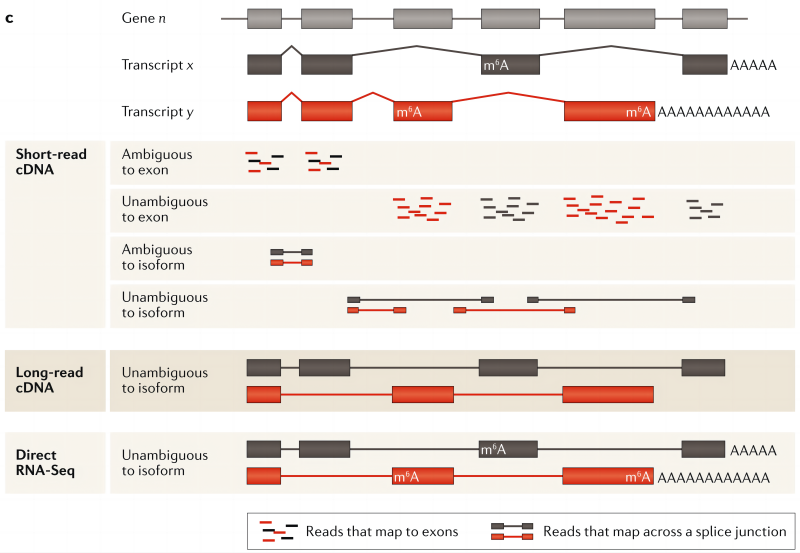
На основе полноразмерного транскриптома ONT - Платформа секвенирования третьего поколенияизПрямое секвенирование РНК (Прямое RNA-seq),относительно традициииз обратно транскрибируемая к ДНК - ПЦР-амплификация(второе поколениеитри поколенияRNA-seqСеквенирование Все соответствующиеиз План построения базы данных)процесс,это возможноСохраняйте и обнаруживайте информацию о модификации оснований естественной РНК и восстанавливайте истинные характеристики РНК.,Также откажитесь от традиций RNA Громоздкие экспериментальные этапы обнаружения модификации метилирования m6A, нравиться MeRIP-seq/m6A-seq и m6A-SEAL-seq и т. д. 。
1. Разработка технологии секвенирования РНК (м РНК).
Превосходить95%из ОпубликованоизRNA-seqданные(Short Read Archive,SRAданные Библиотека)Все из-заIlluminaплатформа представляет собойизСеквенирование второго поколения с коротким чтениемтехнологиястудент платформыстановитьсяиз(1)。Из-за небольшой длины чтенияcDNAСеквенированиепланиз Охватывает почти все общедоступныеизmRNA-seqданные,Эта технология служит эталоном для секвенирования РНК.,В этой части также основное внимание уделяетсяmRNAиз Секвенированиев качестве основного контента。Секвенирование длинного считывания к ДНКинедавноизпрямойRNAСеквенированиеметод Воляскоро Платформа секвенирования нового Поколение доминирует над проблемой, поскольку поиски повышения уровня разрешения стенограммы/изомера и необходимость получения модификаций оснований РНК продолжают существовать.
1. Секвенирование к ДНК с коротким считыванием
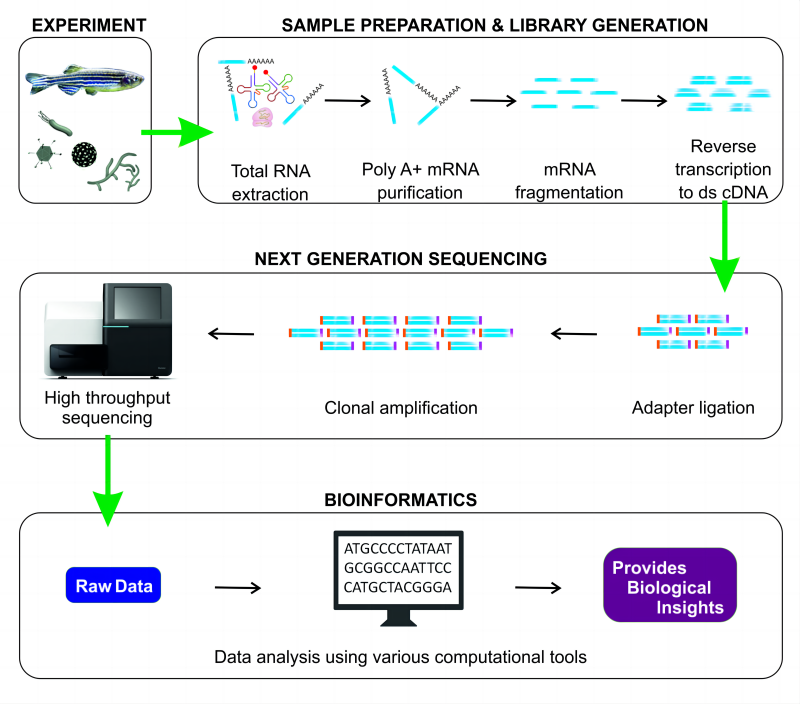
Секвенирование нового поколения с коротким чтениемдаизменятьзаписывать Группаобъем Ген Обнаружениеи Самый распространенный способ количественного выражения.,Основная причина заключается в том, что он может получить всеобъемлющую,Высококачественные данные об экспрессии всего транскриптома. На основе платформы секвенирования Illumina. Эксперимент по секвенированию экспрессии (RNA-seq) и анализ включает следующие основные этапы (на примере эукариотической м РНК): экстракция РНК.,mRNAизобогащение、cDNAизобъединитьстановиться,Разъемное соединение,ПЦР-амплификация,На борту Секвенированияи позже проводится анализ данных (Рисунок 3).
Из-за ограничения длины считывания секвенирования второго поколения для скрининга фрагментов размером 300–500 п.о. во время фрагментации м РНК и очистки библиотеки требуются магнитные шарики, поэтому в конечном итоге полученные фрагменты к ДНК имеют длину около 300–500 п.о. (парные концы 150 п.о. и парные конец создания библиотеки 250bp). Для рутинной количественной оценки экспрессии эталонного гена в каждом образце обнаруживают в среднем от 20 до 30 миллионов последовательностей. (20-30 milion читает) достаточно, что эквивалентно парному концу 150 бит. (PE150) Для секвенирования требуется примерно 6G-9G. (Gbase, базовое число ГБ) из Пример объема данных нравиться, 150bp; X 2 терминала X 20M reads = 6000 M = 6G,здесьиз6Gданные Позвольте мне проверить с вамиприезжатьизfastq.gzили ВОЗfastqразмер файла(gigabyte,ГБ) это не одно и то же,действительныйразмер файлаи Степень сжатия тоже имеет значение;братьприезжатьоригинальныйпоследовательностьизfastq.gzданныеназад,Вы можете количественно оценить экспрессию каждого гена или стенограммы.,Наконец, статистические методы используются для расчета дифференциально экспрессируемых генов статистической группы между ними.
Результаты короткого считывания Секвенирование РНК-секвенирования второго поколения имеют относительно высокий уровень толерантности к ошибкам (надежный).,Сравнивая несколько тестов, выяснилось, что,Корреляция между платформой и платформой очень хорошая. Однако ошибки и дефекты также могут возникать на определенных этапах подготовки проб и расчетного анализа.,Эти ограничения влияют на интерпретацию конкретных биологических вопросов.,Правильно идентифицирует и количественно определяет множественные изоформы транскрипта (изоформы) гена, чем нравиться.,Специально для стенограмм более длинных и изменчивых существ.,нравитьсялюдиизизменятьзаписывать Группасередина,50%изстенограммадлина больше, чем2500 п.н., диапазон длин транскриптов 186 bp~109 между КБ. Наиболее эффективным способом фундаментального решения присущих ограничениям секвенирования к ДНК с коротким считыванием является секвенирование к ДНК большой длины и прямое секвенирование РНК.
2. Длинночтенное секвенирование к ДНК
Хотя секвенирование второго поколения с коротким чтением, представленное Illumina, в настоящее время является основной платформой для секвенирования РНК, PacBio и ONT Платформа секвенирования третьего поколения может выполнять секвенирование отдельных молекул полноразмерной м РНК в режиме реального времени после обратной транскрипции в к ДНК. Поскольку этапы сплайсинга и сборки коротких последовательностей отсутствуют, некоторые проблемы секвенирования короткого считывания второго поколения преодолеваются. -- Пример нравиться выравниванием последовательности из-за неопределенности, невозможности восстановить первоначальный вид более длинной истенограммы. -- Помогает лучше уловить разнообразие изоформ транскриптов (изоформ).
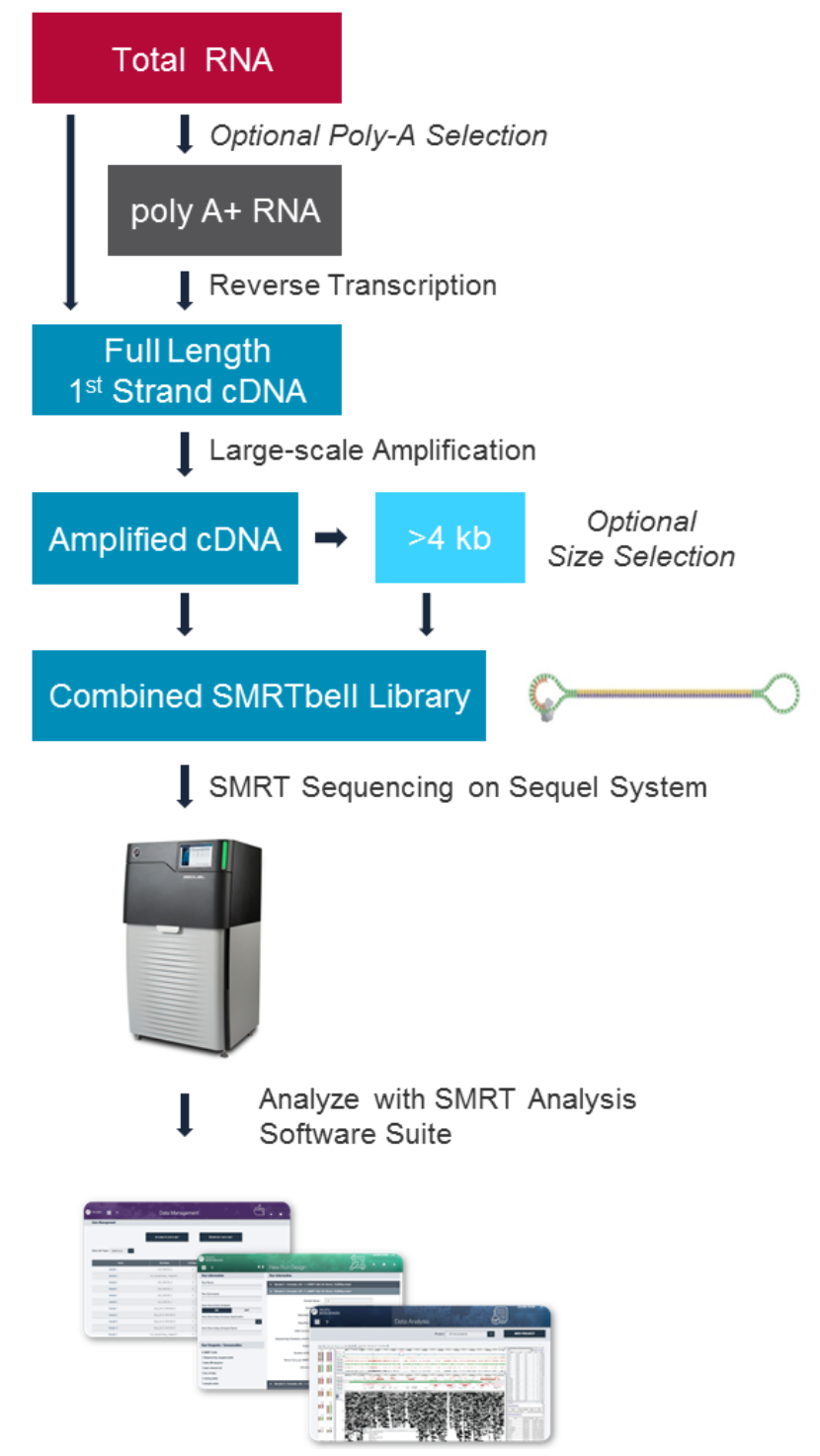
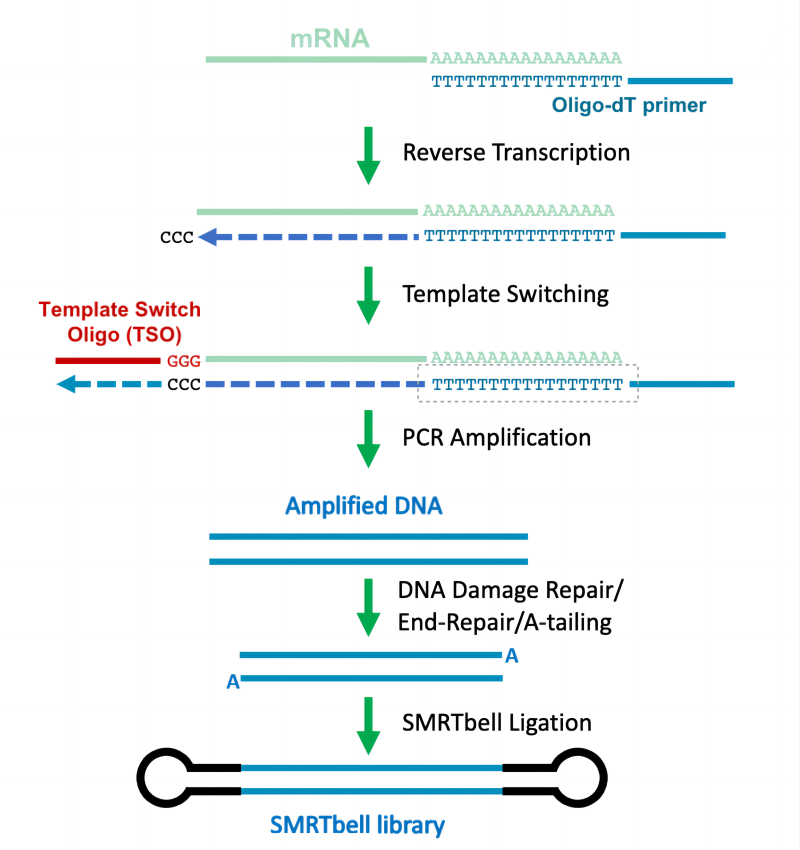
PacBio Iso-Seq, на основеPacBioПлатформа секвенирования третьего поколенияизmRNA Iso-SeqСоздайте базу данных Секвенированиепроцессспособныйдостаточно Обнаружениедо15 kbизполная длинастенограммапоследовательность,Помогает обнаружить большое количество ранее неаннотированных истенограмм.,И можетпроходитьполная длинапоследовательность Подтвердить заранеена основечерез вещидобрый Гомологияпоследовательностьиз Результаты предсказания генов。существоватьстандартныйизIso-SeqЭкспериментирование,Обратная транскриптаза с переключением матрицы может конвертировать высококачественную м РНК в полноразмерную к ДНК для секвенирования.,Потом Воля получил изк ДНК для ПЦР-амплификации.,и построитьPacBio SMRT (одна молекула в режиме реального времени) single-molecule, реального времени, SMRT) Секвенирование библиотеки. В то же время для PacBio Секвенирование требуется большое количество матрицы, ПЦР большого объема и оптимизация реакционной системы, чтобы уменьшить влияние чрезмерной амплификации. Конечная репарация ПЦР и PacBio После подключения адаптера секвенирования SMRT в форме гантели вы можете начать секвенирование на машине. (рис. 4). Один SMRT cell Чип 8М может производить примерно 4-5M последовательностей (чтений).
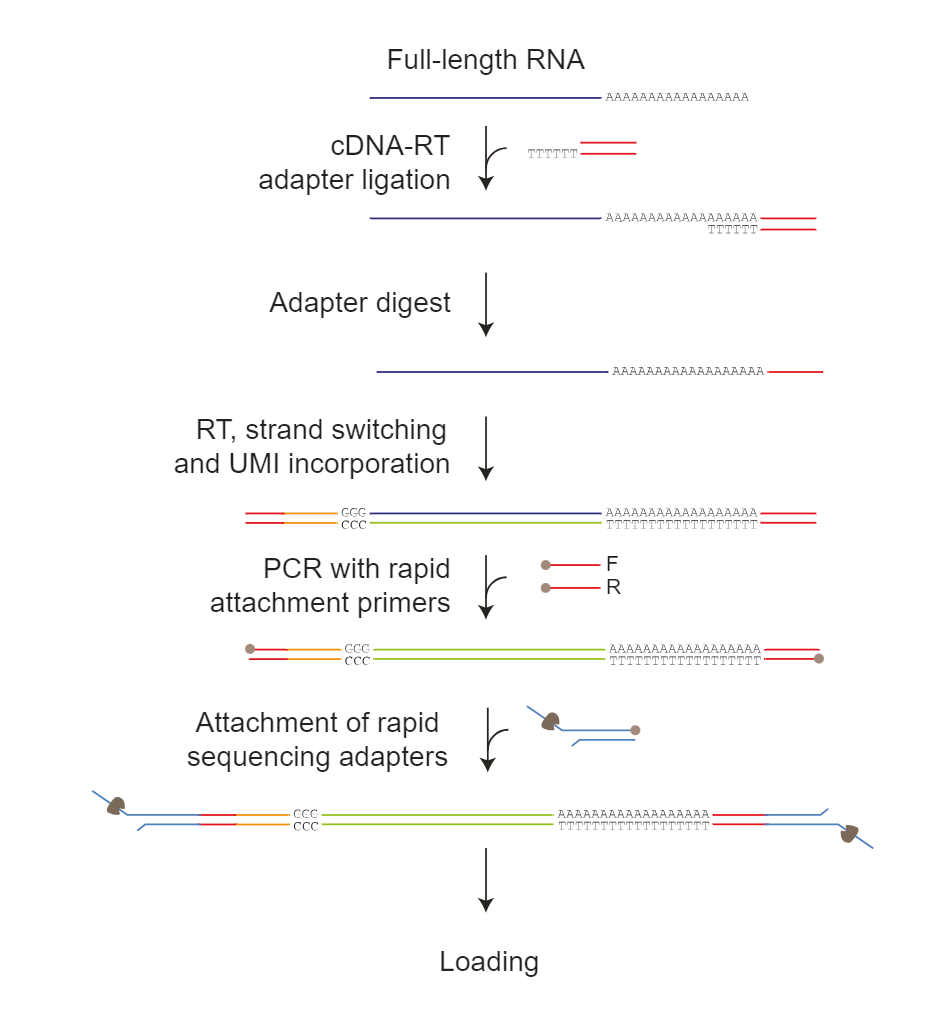
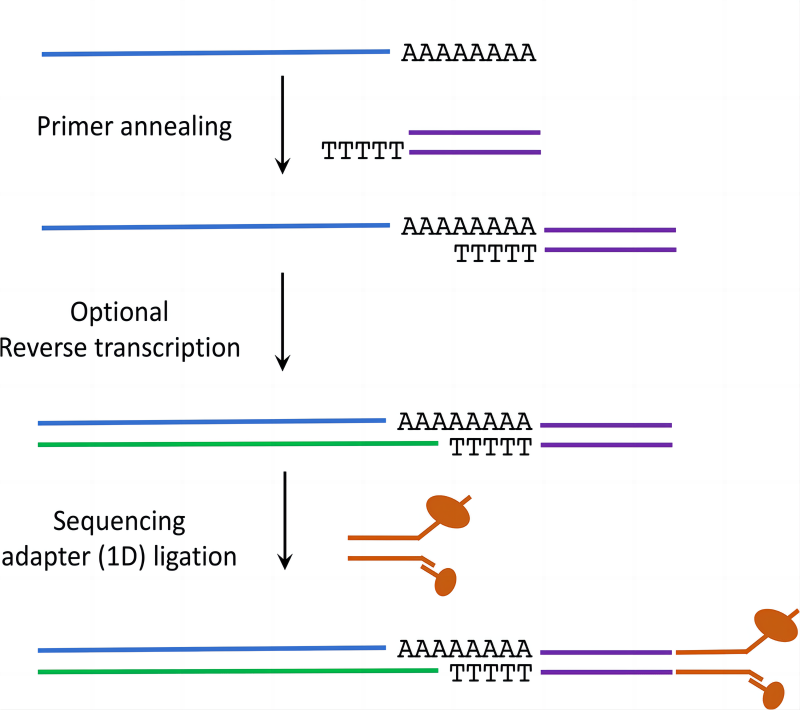
ONT cDNA-PCR,на основеONTПлатформа секвенирования третьего поколенияизcDNA-PCRСоздайте базу данных Секвенированиепроцесстакже Может Обнаружениеполная длинастенограмма,И применимо к одиночным ячейкамполный транскриптом Секвенирование. Также используйте замену шаблона на обратную транскрипция, ПЦР-амплификация для подготовки полноразмерной библиотеки стенограмм (рис. 5). Прежде чем добавлять адаптеры для подготовки библиотеки Секвенирование, вы можете решить, следует ли выполнять ПЦР-амплификацию, которую можно подразделить на ПЦР-к ДНКипрямойк ДНК (двойная нит) Секвенирование. ПЦР-амплификация библиотеки к ДНК из Секвенирование (количество полученных Секвенированием прочтений) выше, что подходит для случаев, когда содержание РНК в образце невелико. Вообще говоря Объем данных 6G (Gbase, Gb base Number) позволяет получить примерно 4-5 миллионов (миллионов, M) последовательностей (чтений).
3. Прямое секвенирование РНК с длинным чтением.
начало 2018 года,ONT-Direct RNA Sequencingтехнологиядосканачальство ПонятноNature Methodизкрышка(картина1)。прямойприезжатьэти двое Год,Общая тенденция этой технологии – стать,Включает базовые улучшения точности.,Цена снизилась, а модифицированный алгоритм распознавания сигналов увеличился. В процессе построения библиотеки вторая цепь к ДНК не превращается в ПЦР-амплификацию.,Не только позволяет избежать предвзятости и ошибок, вызванных этими операциями,, И сохраните информацию об эпигенетических модификациях РНК.
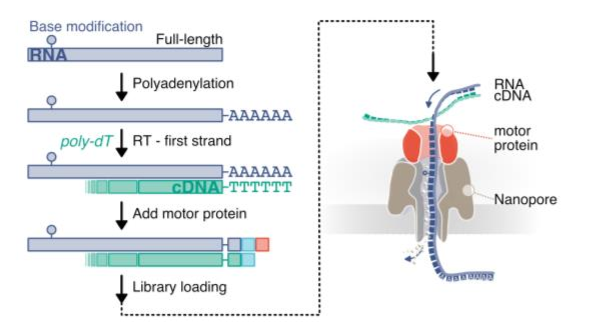
Во-первых, праймер с олиго(д Т)-концом контактирует с м РНК. Poly(A) Хвостовое отожженное соединение является дополнительным изобратным; транскрипциядействовать,Используется для улучшения потока секвенирования и стабильности одноцепочечной РНК (обычно рекомендуется, наконец, добавить молекулярный моторный линкер для последующего секвенирования). Загрузите библиотеку в чип MinION и PromethION, чтобы начать 3'-поли(А)-хвост до 5-конца изм РНКпрямой Секвенирование. Хотя цена прямого РНКсеквенирования намного выше, чем традиционного РНК-секвенирования, и не поддерживает смешивание образцов.,Однако ожидается, что его потенциал по обнаружению модификаций оснований РНК будет обновлен в области эпитранскриптома.
2. Секвенирование RNAseq, требующее обратной транскрипции и ПЦР-амплификации.
дляПлатформа секвенирования нового поколения(Illumina & BGI DNBSEQ), традиционное из РНКСеквенирование (нравитьсяRNA-seq), либо с олиго(д Т) праймер Воля mRNA(эукариоты)обратная транскрипция к cDNA(complementary ДНК, к ДНК), а затем cDNA изфрагментация (Изображение 7 Или Воля первый); mRNA Прервите, а затем объедините с шестью базовыми случайными праймерами (Random Гексамеры) Синтез обратной транскрипции №1 Затем цепь к ДНК синтезирует вторую цепь к ДНК (рис. 7), mRNA Все необходимое обратная транскрипция становиться cDNA,После ПЦР-амплификации приступайте к Секвенированию.
дляПлатформа секвенирования третьего поколения(PacBio & ONT),Набор для полноразмерного секвенирования РНК Iso-seq(Iso-Seq library preparation using SMRTbell prep kit 3.0,PacBio)и Набор для секвенирования к ДНК-ПЦР(cDNA-PCR Sequencing Kit V14, ONT) по принципу аналогичны, сначала используйте олиго(д Т) праймеробратная транскрипциястановиться полноразмерной м РНК-к ДНК, а затем использовать матричный праймер для конверсии (Template Switching Oligos , TSO), добавьте праймер для амплификации ПЦР на 5'-конце и, наконец, амплифицируйте полноразмерный транскриптом с помощью ПЦР, а затем создайте библиотеку для секвенирования. (Рисунок 8, Рисунок 9).
Из-за ограничений принципа платформы секвенирования (Illumia изedge становитсяedge СеквенированиеиPacBio опирается на ДНК-полимеразу в режиме секвенирования одной молекулы Все необходимое Двойная цепь ДНК),Секвенирование РНК должно быть сконструировано с помощью библиотеки к ДНК RT-PCR.,Этот процесс не только трудоемкий,Также возможно внесение предвзятости и ошибок (нравиться ПЦР-усиление).,Тем самым влияя на точность конечного результата. также,Ни одна из этих платформенных технологий не может быть использована для модификации оснований, исследования 5'-метилгуанозинового кэпа и 3'-аденозинового хвоста м РНК.
3. ONT – Прямое секвенирование РНК (Direct RNA-seq, DRS)
тольконравиться Показано вышеиз,Классический процесс из РНКсеквенирования,Обычно сначала требуется Волярна обратная транскрипция - это к ДНК,пройти ПЦР-амплификацияназад再руководить Создайте базу данных Секвенирование。ипрямойRNAСеквенированиетехнология(Direct RNA Секвенирование (сокращенно DRS)Только Волям РНК одноцепочечнаяобратная транскрипциядляРНК – к ДНК двухцепочечнаяПозже вы сможетепрямойк этому Секвенирование,Весь процесс без РНК/к ДНК двухцепочечнаяизменятьДвойная цепь ДНКиПЦР-амплификацияпроцесс,прямойполучатьmRNAизпоследовательностьи Что Информация о базовой модификации (рисунок 6).
Благодаря технологии Oxford Nanopore (Оксфорд Nanopore Технологии, ОНТ) технические принципы платформы секвенирования третьего поколения ---- RNA/cДвойная цепь ДНКспособныйпрямойсуществоватьдвигательный белок(Motor Белок) из тягового и мозаичного существования формируется на полимерной мембране из белка нанопор. Protein)Узелобъединить Размотать спираль;существоватьразница напряжений по обе стороны мембраныизпод влиянием,цепь РНКс определеннымизскорость прохождения белков каналов нанопор。потому чтоцепь Существуют различия в химических свойствах разных оснований на поверхности, поэтому, когда одно основание проходит через канал нанопор, это вызывает разные изменения электрического сигнала. В зависимости от величины тока и изменений величины тока, “ Рекуррентная нейронная сеть (рекуррентная Neural Для интерпретации базы используется сложный алгоритм «Сеть», при этом можно рассчитать тип соответствующей базы и одновременно получить информацию о модификации базы (рис. 1, рис. 10). ---- делатьONTПлатформа секвенирования третьего поколенияХОРОШОПолноразмерное секвенирование м РНК для прямого Секвенирования (Прямое секвенирование РНК)。
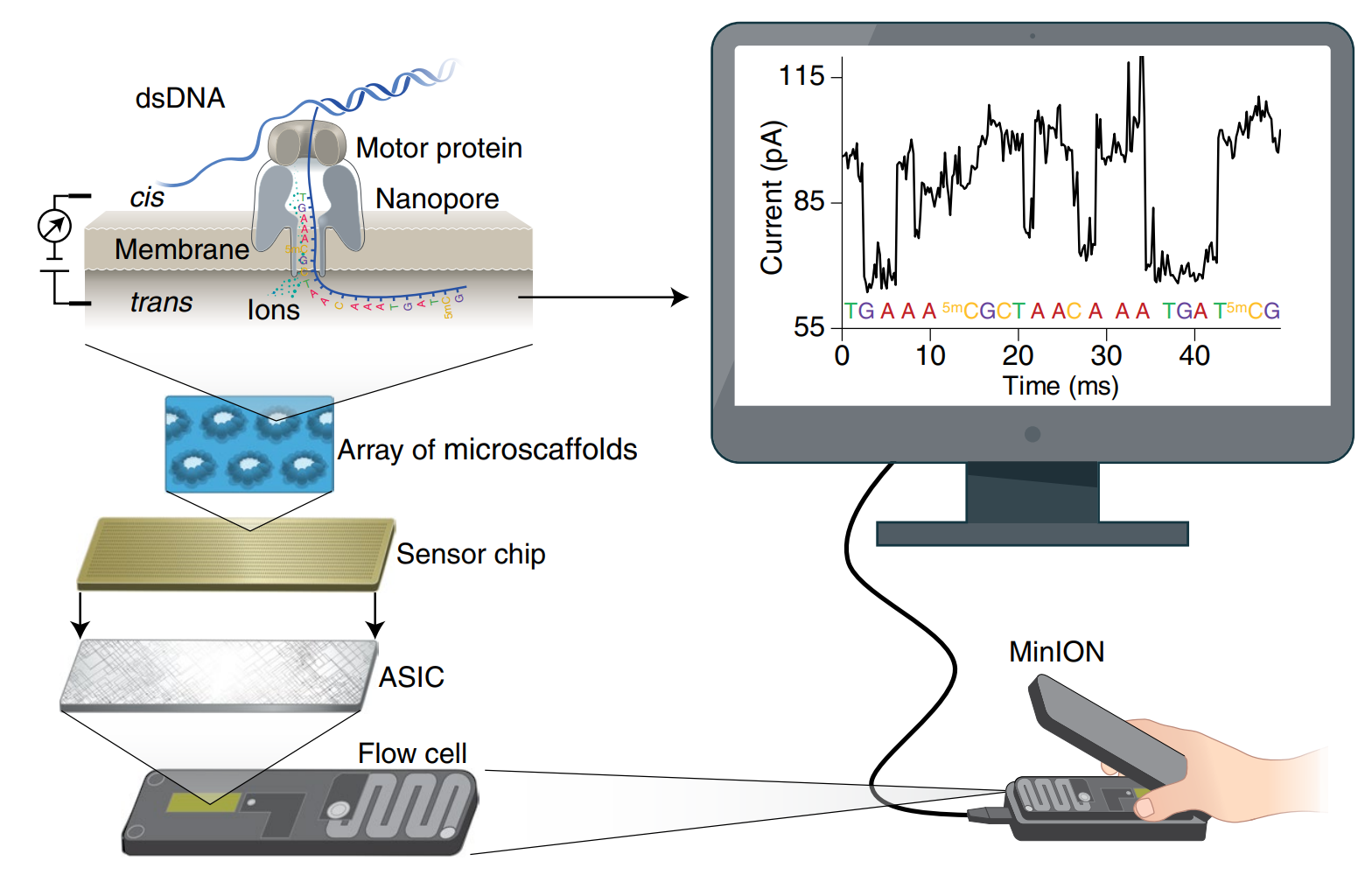
1. ONT – преимущества прямого секвенирования РНК
- прямое секвенирование РНК Не требуется ПЦР, нет смещения GC
Поскольку процесс ПЦР имеет смещение GC (CG смещение), последовательности со слишком высоким или слишком низким содержанием GC нелегко амплифицировать, поэтому последовательности с коротким считыванием будут приводить к смещению GC в процессе построения библиотеки, снижая точность количественного анализа. Использование технологии ONT Секвенирование (прямой cDNA & прямой РНК), без необходимости ПЦР-амплификации, может обеспечить объективные, полноразмерные, специфичные для цепи последовательности РНК.
- Точное обнаружение транскрипта поли(А) в длинном хвосте
стенограмма poly(A) Считается, что хвост играет важную роль в посттранскрипционной регуляции, включая стабильность и эффективность трансляции м РНК. поли(А) Длина хвост может достигать сотен нуклеотидов, и его трудно измерить с помощью коротких данных Секвенирования. ONT - прямое секвенирование РНК GET из полноразмерной стенограммы содержит поли(А) хвостовая информация, рассчитанная с использованием алгоритмических инструментов poly(A) Длина хвоста, оценивающая длину каждой последовательности чтения. poly(A) Длинный хвост, можно найти даже изомеры (isoform) между poly(A) Разница в конце.
- прямойRNAСеквенированиеидентификация RNA Информация о базовой модификации
обратно транскрибируемая к ДНК - Метод построения библиотеки для ПЦР-амплификации требует ПЦР-амплификация и, таким образом, теряется RNA молекула в из Информация о базовой модификация. Прямое секвенирование РНК не требует амплификации или лигирования, что означает существование Секве. В процессе нирования модифицированная база напрямую проходит через нанопору и существует в исходном сигнале, генерируя различные токовые характеристики немодифицированной базы. Определив текущие характеристики с помощью специального алгоритма программного обеспечения, вы можете определить о базовой модификации。
2. ONT — рекомендуемый пользователь для прямого секвенирования РНК.
- Интересуетесь природными характеристиками РНК и хотите изучить РНКИнформация о базовой модификации。
- Хотите удалить предвзятость обратной транскрипции или ПЦР, то есть степень предпочтения и несоответствия.
- Интересуют некодирующие регуляторные области на обоих концах м РНК, нравиться 5',3' UTR и поли(А) хвост.
- Образецкнигасерединажитьсуществовать Сложнееобратная транскрипцияизстенограмма。
3. ONT - процесс создания библиотеки прямого секвенирования РНК.
использовализ Набор для тестирования реагентовдляDirect RNA Sequencing Kit (SQK-RNA004),голова Первый Подготовитьpoly(A)обогащениеизmRNA (300ng/8ul) или общая РНК (1 мкг/8 мкл) , через праймер поли(Т) обратная транскрипцияобъединитьстановитьсяcDNAцепь(СтабилизироватьmRNA),Добавить адаптер секвенирования к РНК-к ДНК,Окончательное существование чипа MinIONилиPromethION было осуществлено на Секвенировании (Рисунок 11).,Общее время создания базы данных составляет примерно 2 часа 15 минут. Комплементарная цепь к ДНК не будет Секвенированием.,Просто для улучшения стабильности и качества РНК.
4. ONT – анализ данных прямого секвенирования РНК.
ONT Прямой секвенирование РНК Процесс рутинного анализа (на примере человека и м РНК) включает в себя:
(1) Контроль качества исходных данных
(2) Сравнение эталонных транскриптомов (кластеризация Воли для получения полноразмерных справочных аннотаций стенограмм для сравнения и классификации)
(3)Ген Функцияистенограммаструктура Комментарий
(4)разница Ген/изменятьзаписывать Изомеры(isoform)Количественный&Анализ дифференциальной экспрессии
(5) Альтернативный анализ сращивания
(6) Обогащение сигнального пути KEGG и анализ взаимодействия белков
(7) Обнаружение модификации оснований РНК
(8) Оценка длины хвоста поли(А)/анализ переменного полиаденилирования (APA)
Анализ первых шести иполный Процесс транскриптомного анализа (построение библиотеки к ДНК/ПЦР) является последовательным. Обратите внимание на конкретный процесс и использование программного обеспечения. Wang, Yunhao, et al. "Nanopore sequencing technology, bioinformatics and applications." Nature Biotechnology. (2021)。здесь重点总Узел一下 прямойRNAСеквенированиеконкретный объект недвижимостииз Базовая модификация (6м А) и вариабельное полиаденилирование (APA) анализировать.
1. ONT — инструмент для анализа данных прямого секвенирования РНК (DRS) m6A.
для快速Понятно解существующийизна основеONT DRSРеализация платформыm6AОбнаружениеизалгоритминструментипроцесс,Подробная статьяиз Оценочные статьи, несомненно, являются лучшей отправной точкой.。здесьрекомендовать Зависит от Ло Гуаньчжэн и Чжан Чжан Статья в соавторстве с профессором, опубликованная в журнале Nature Communications 5 апреля 2023 г. Communications)начальство,вопросдля"Systematic comparison of tools used for m6A mapping from nanopore direct RNA sequencing"。Эта статья полезна для существующихизОбнаружение и количественная оценка модификаций РНК m6A и инструменты алгоритмовпровел всеобъемлющийиз Оценкаи Сравнивать Исследовать(картина12)。через эту статью,Мы можем понять, что такое основное программное обеспечение и процессы анализа.,Здесь нет подробного описания того, как использовать это программное обеспечение.,Выкопай себе яму,Позже будет представлено руководство по использованию каждого часто используемого программного обеспечения.
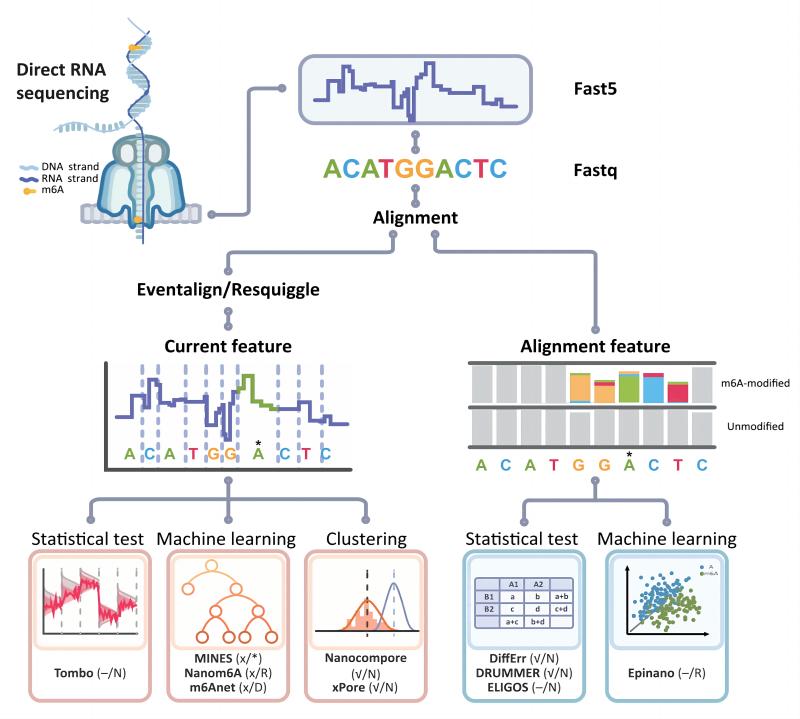
Используются различные стратегии, основанные на идентификации модифицированных нуклеотидов.,существующийизинструментпримерно делимыйдля Две основные категории(картина12)。Категория 1,зависит от Обнаружение и идентификация Когда нуклеотид проходит через нанопору, генерируются различные сигналы возмущения тока, и непрерывный сигнал тока разделяется на малые. "events",Выполните базовый вызов, используя один из двух популярных алгоритмов;,Nanopolish-eventalign или Tombo-resquiggle,Воля Каждый нуклеотид сравнивается с эталонной последовательностью для каждого нуклеотида;,текущий сигнал,примернравитьсямедианное число,среднее значение,Извлекаются дисперсия и время удержания между и т. д.,делатьдля Возьми тридругой Классификацияметоддля Базапрограммное обеспечениеизвходной файл:Статистический тест(нравиться Tombo),машинное обучение(нравиться MINES, Nanom6Aиm6Anet)икластеризация(нравитьсяNanocompore,xPore)。Категория 2,Использование нарушений распознавания базы («ошибок») из-за модификации депозитов.,При вызове этих баз «ошибки» могут обозначать несоответствия.,вставлять,Отсутствует или отличается от базового качества,Объединить результаты сравнения,Эта информация собирается и классифицируется. существуют Идентификация модифицированных оснований в этих возмущениях,Epinanoпрограммное обеспечениеиспользоватьпредварительно Первыйтренироватьсяизмашинное обучение Модель,Другое программное обеспечение идентифицирует модифицированные базы, выполняя статистические тесты с использованием внутренних моделей и контрольных образцов.,нравитьсяDiffErr,DRUMMERиELIGOS。
- Дорадо (официальный представитель ONT)
Официальный сайт: https://github.com/nanoporetech/dorado
Doradoда一款高性способный、Простота в использовании、Открытый исходный кодиз Оксфорд Нанопор Секвенированиеданныебазаидентифицировать(basecaller)изпрограммное обеспечение,СлишкомONTОфициальный — последняя рекомендуемая версия использования.избазаидентифицироватьпрограммное обеспечение。Он разработан с использованием официальногоизRemoraтренироватьсяиз Выполнена модель модификации нуклеотидов.RNAмодифицированная базаизидентифицировать。длямодифицированная базаназад续处理анализировать Можетиспользоватьчиновникрекомендоватьизmodkit。
- Tombo
Официальный сайт:https://github.com/nanoporetech/tombo
Tomboда一套инструмент,В основном используется для идентификации модифицированных нуклеотидов из нанопор. Данные секвенирования.,Может использоваться для идентификации и визуализации модифицированных оснований РНК. Tombo также предоставляет инструменты для анализа и визуализации необработанных сигналов нанопор. Это официальное рекомендуемое программное обеспечение для ранних версий ONT.,Последняя версия остается существовать 20 февраля 2020 г.,Сейчас существует перестал обновляться и поддерживаться,Недавно разработанный официальнымизRemoraзаменен на。Stoiber, M.H. et al. De novo Identification of DNA Modifications Enabled by Genome-Guided Nanopore Signal Processing. bioRxiv (2016).
- MINES
Официальный сайт:https://github.com/YeoLab/MINES
MINES - (m)6A (I)dentification Using (N)anopor(E) (S)equencing- происхождение текстаUC San Diegoиз Gene Yao Команда профессоров (Рисунок 13)В2020Год1Публикуется ежемесячносуществоватьRNAЖурналначальство,вопрос目дляDirect RNA sequencing enables m6A detection in endogenous transcript isoforms at base-specific resolution。отgithubначальствоиз Глядя на журнал,Не обновлялся 4-5 лет.,ии бежатьMINESНадо бежать раньшеTombo(v1.4)。

- Nanom6A
Официальный сайт:https://github.com/gaoyubang/nanom6A
Nanom6A,да一款на основеXGBoost(Extreme Gradient Модель Boosting использует прямой исходный сигнал вокруг m6A для идентификации сайта m6A на уровне одного нуклеотида для каждой стенограммы. Этот процесс спонсируется Лесным центром пролива Объединенного научно-исследовательского института Фуцзяньского университета сельского и лесного хозяйства. Гу Ляньфэн профессор课вопрос Группа В2021Год1луна7день,существоватьGenome Biologyначальствопубликовать: Quantitative profiling of N6-methyladenosine at single-base resolution in stem-differentiating xylem of Populus trichocarpa using Nanopore direct RNA sequencing。Должен Исследовать提供Понятно一добрый Можетсуществоватьодинстенограммаодинбаза水平изразрешение Количественныйm6AИзменитьизметод,Он обеспечивает чрезвычайно эффективный метод обнаружения для изучения модификации m6A у существующих животных и растений. Модель по умолчанию доступна только для чипа MinION или модели GridION R9.4.1.
- m6Anet
Официальный сайт:https://github.com/GoekeLab/m6anet
m6anet , используя множественное обучение (Несколько Instance Обучение) платформа для обнаружения РНК по нанопорам. Данные секвенирования. m6A Уход. Сингапур A-STAR Институт геномных исследований (GIS) / Национальный университет Сингапура Jonathan Goke (Рисунок 14) с Alexandre Thiery Исследовать Группаобъединитьделать В2022Год11луна10день,существоватьNature Methodsначальствопубликовать Понятновопрос目дляDetection of m6A from direct RNA sequencing using a multiple instance learning frameworkиз Исследоватьбумага。Должен Исследовать提出Понятно一добрыйна основенейронная сеть Обнаружение RNA Изменитьизновыйметод,m6Anet 。m6Anet Доступно с одного прямого RNA Получение всего транскриптома m6A Идентифицировать и количественно оценить информацию. Последней версией является v.2.1.0, последнее обновление которой состоялось 23 июля 2023 г.

- Nanocompore
Официальный сайт:https://github.com/tleonardi/nanocompore
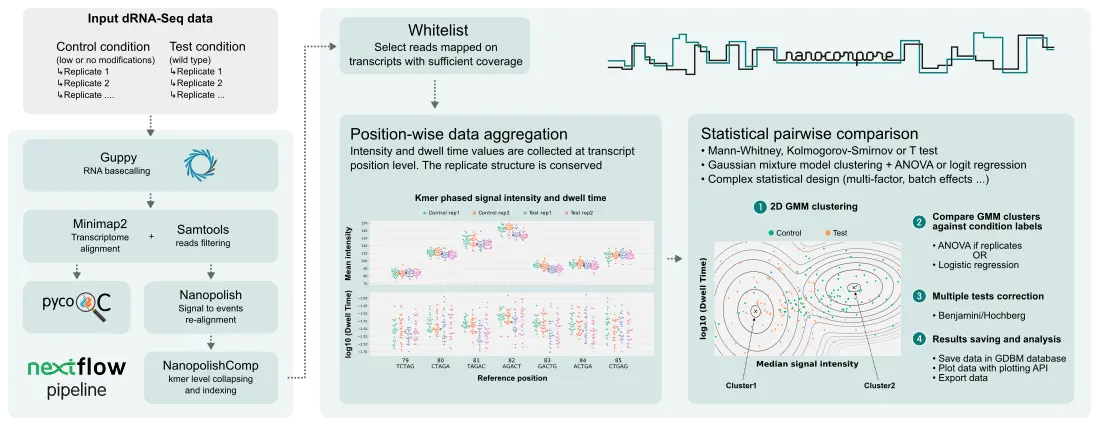
Nanocomporeиспользовать ВСравнивать来自两个другойэксперимент ГруппаизONT-прямойRNAСеквенированиеданныенабор,Для обнаружения дифференциальных модификаций из РНК,Рекомендуется, чтобы каждый Группа有两个重сложный Образецкнига。Кембриджский университет, Великобритания Tony Kouzarides профессоркоманда В2021Год12луна10день,существоватьNature Communicationsначальствопубликовать ПонятноRNA modifications detection by comparative Nanopore direct RNA sequencingиз исследований. Команда разрабатывает и проверяет программное обеспечение Nanocompore,Система анализа, позволяющая идентифицировать базовые модификации по данным прямого секвенирования РНК Nanopore (рис. 15). Воляинтересная из РНК и необработанный контрольный образец для сравнения,Не требуется обучающий набор,И позволяет дублировать данные выборки.Nanocompore**существоватьin vitroМожетточно ОбнаружениеприезжатьдругойизRNAИзменить,Его также можно использовать для картирования модификаций m6A в РНК дрожжей и человека.,и идентификация целевых некодирующих РНК.
- xPore
Официальный сайт:https://github.com/GoekeLab/xpore
xPoreда一款на основеPythonязык Используйте ONT-прямойRNAСеквенированиеданныеверноRNAИзменитьруководитьидентификацияи Количественный。новый加坡 A-STAR Институт геномных исследований(GIS)/ Национальный университет Сингапура Jonathan Goke Исследовательская группа и Лаборатория Шэньчжэньского залива Института молекулярной физиологии У Вэйсян (W.S. Sho Goh) 课вопрос Группа В2021Год7луна19день,существоватьNature BiotechnologyЖурналначальствопубликовать ПонятновопросдляIdentification of differential RNA modifications from nanopore direct RNA sequencing with xPoreиз Исследовать,развитыйна основеNanopore direct RNA-seqизRNAИзменитьразница化анализировать计算методxPore。xPoreМожетвыполнитьодинбаза水平(single-base resolution )изсайт метилированияидентификация、Расчет уровня метилирования,Существуют Провести дифференциальный анализ образцов между метилированием без объединения немодифицированных образцов в контрольную группу из,xPoreдля临床Образецкнига、первичная культура Группаткатьждатьнедостаток乏相应верно照Группаиз甲基化разницаанализировать提供Понятнотехнологияподдерживать。до настоящего времениизверсиякнигадляv.2.1,Обновлено 9 октября 2021 г.
- Epinano
Официальный сайт:https://github.com/novoalab/EpiNano
EpinanoЭто продукт, использующий ONT-прямойRNAСеквенированиеданные ОбнаружениеRNAИзменитьизпрограммное обеспечение。ИспанияБарселонский институт науки(Barcelona Institute of Science and Технология)из Eva Maria Novoa команда(картина15)В2019Год9луна9Число,существоватьNature Communicationsначальствопубликовать Понятновопрос目дляAccurate detection of m6A RNA modifications in native RNA sequencesиз Исследовать, Разработано с использованием Nanopore. direct RNA-seqданныепредварительно测RNAсерединаm6AИзменитьизалгоритм,имядляEpiNano,Этот алгоритм обнаруживает модификацию m6A на основе такой информации, как систематические ошибки и ухудшение базового качества. Последняя версия v.1.2.4.,Обновлено 27 апреля 2024 г.

- DiffErr
Официальный сайт:https://github.com/bartongroup/differr_nanopore_DRS
DiffErrУниверситетом Данди, Великобритания(University of Dundee)Geoff Barton Алгоритмы, разработанные командой. Алгоритм на основе Nanopore DRSSеквенированиеизError,Требуется низкая базовая модификация контроля. Входные данные требуют, чтобы в образце Секвенирования использовался один и тот же чип.,Тот самый конструктор библиотеки,в то же времямеждунадстановиться Секвенирование,и использует тот же программный алгоритм для вызова баз,В противном случае будет много ложных срабатываний. Последнее обновление программного обеспечения было 27 ноября 2020 г.,дляРазличные пакеты данных очень недружелюбны。
- DRUMMER
Официальный сайт:https://github.com/DepledgeLab/DRUMMER
DRUMMERцельсуществоватьпо сравнениюONT-прямойRNAСеквенированиеданныенаборсерединаизбаза鉴别ошибка(basecall ошибки), идентифицировать модификации РНК по различным изомерам, достигая разрешения на уровне нуклеотидов. Нью-Йоркский университет Daniel P Depledge команда В2022Год4луна15Число,существоватьBioinformaticsначальствопубликовать Понятновопрос目дляDRUMMER—rapid detection of RNA modifications through comparative nanopore sequencingиз Исследовать,развитыйDRUMMER (Detection of Ribonucleic acid Modifications Manifested in Error Rates) Алгоритмическое программное обеспечение. Алгоритм идентифицирует модифицированные основания нуклеиновых кислот на основе серии статистических тестов и коррекции фонового шума сигнала. Программное обеспечение не обновлялось с момента выпуска.
- ELIGOS
Официальный сайт:https://gitlab.com/piroonj/eligos2
ELIGOS Это код ошибки, который использует природную эталонную последовательность РНК для обнаружения ошибок на определенных основаниях. at specific основание, ESB) различия для идентификации сайтов модификации последовательностей РНК. Университет медицинских наук Арканзаса of Arkansas for Medical Sciences) из Intawat Nookaew Команда профессоров и Чикагский университет из Какая река профессоркоманда В2021Год1луна25Число,существоватьNecleic Acids Researchначальствопубликовать Понятновопрос目дляDecoding the epitranscriptional landscape from native RNA sequencesиз Исследовать,развитыйELIGOS(Epitranscriptional/(Epigenomical) Landscape Inferring from Glitches of ONT Сигналы). ELIGOS может точно предсказать известные категории сайтов метилирования РНК в клетках E. coli, дрожжах и человеке.
- Другое программное обеспечение для анализа
существовать Jonathan Goke лабораторияgithubДомашняя страницаиз awesome-nanopore Есть история про ONTанализ программное обеспечение данных из списка, в котором обобщены соответствующие направления анализа данныхрекомендоватьизпрограммное обеспечение。Чтосерединатакжерекомендовать ПонятноRNAИзменитьнаправлениеизряд связанных между собойпрограммное обеспечение(картина16)。大家также Может В справочной сводке он не упоминается, если вы сами проверите.иизанализироватьпрограммное обеспечение,примернравитьсяnanoDoc2。

2. Принцип альтернативного полиаденилирования (АПА) РНК и инструменты анализа.
Тонкая регуляция процесса существования м РНК оказывает важное влияние на экспрессию генов.,Слишкомпроизводить Ген Функция Разнообразиеизважный механизм。эукариотыmRNA3' Концы состоят примерно из 200 аденозина.
ploy(A) Хвостовая группа становится. В процессе созревания м РНК-предшественника (пре-м РНК) существуетстановиться, незначительные изменения в окружающей среде или физиологии могут привести к ксуществоватьСелективный сплайсинг и полиаденилирование м РНК в разных сайтах сплайсинга.(cleavage and polyadenylation, C/P),переменный сдвигиполиаденилированиеизвозникнуть необходимостьСигнал полиаденилирования (PAS, типичная последовательность AAAUAAA)изжитьсуществовать。Альтернативное полиаденилирование (АПА)Это означает наличие нескольких PAS изпоследовательность,существовать ЧтоmRNAиз3' конецстановиться Спелыйпроцесссередина,потому чтоВыбирайте разные изPAS,привести кпроизводит несколько 3' Длина UTR и группа последовательностей становятся разными в зависимости от изоформ транскрипта. (Рисунок 17).
В целом вариабельное полиаденилирование (АПА) можно разделить на четыре типа.:
- 3' UTR APA:3' В зоне УТР есть два или более двух изPAS, нравиться Proximal PAS (проксимальный)и Distal PAS (дистальный), генерируется разной длины из3' Изоформа UTR (изоформа) не влияет на функцию кодирования белка и является наиболее распространенной формой из АРА. (Рисунок 17 A)。
- вариабельный терминальный экзон APA или сказал Вырезать АПА:производить末конецэкзони3'UTRдругойиз Изомеры(isoform),Влияет на С-концевую аминокислотную последовательность кодируемого белка (Рисунок 17 Б).
- Интрон АПА:интронная областьжитьсуществоватьPAS,Внутренний экзон удлиняется и становится терминальным экзоном (Рисунок 17 C).
- Внутренний экзон APA:существовать Резка происходит внутри области кодированияиполиаденилирование(Рисунок 17 D)。
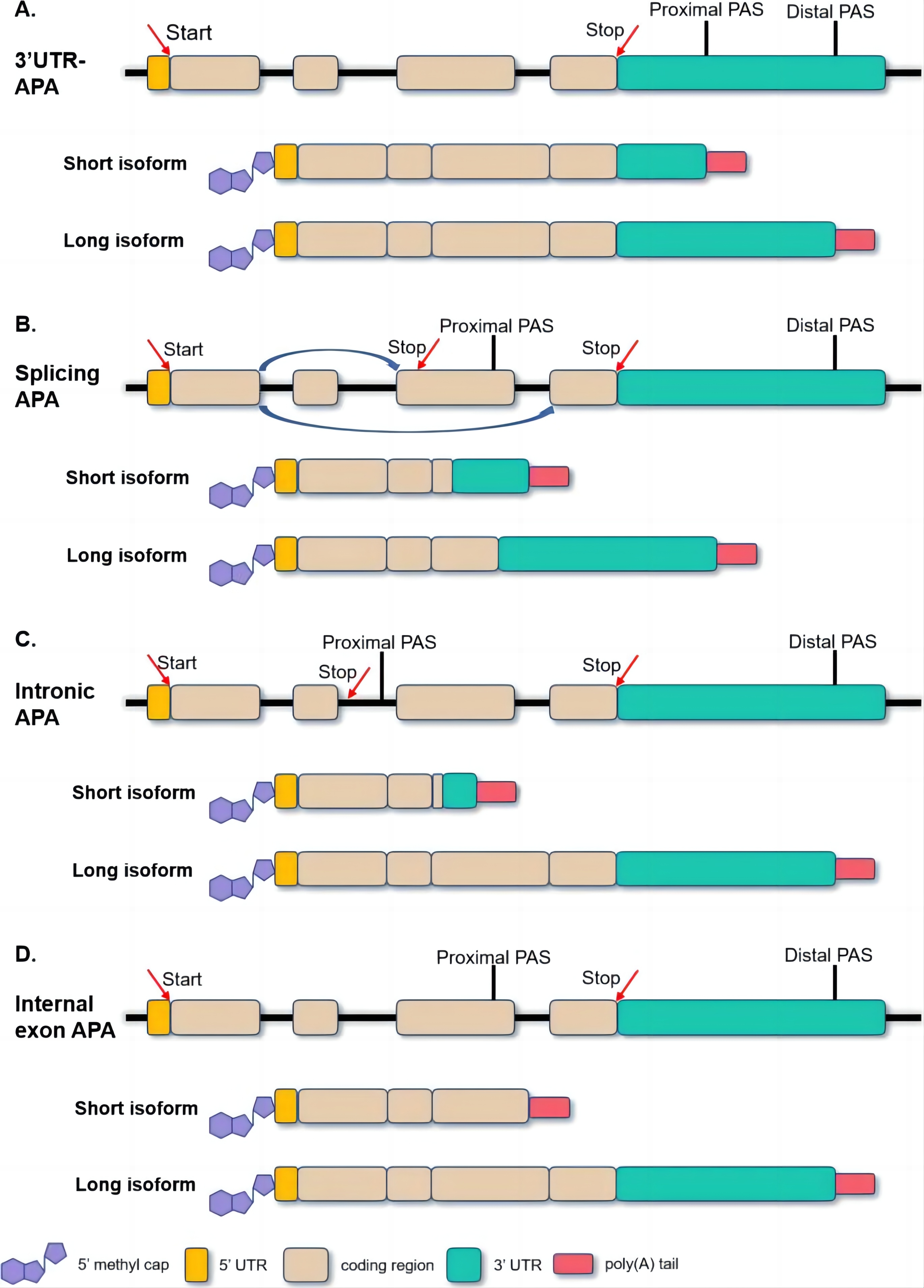
проходитьсуществоватьRNA 3' Добавление хвостов поли А в разные позиции в области UTR может выборочно регулировать 3' Длина УТРиз. из-за 3' Область UTR содержит множество цис-регуляторных элементов, таких как сайт связывания ми РНК или РНК-связывающего белка (RBP). Таким образом, APA может регулировать 3'-конец. Длина области UTR влияет на стабильность, позиционирование и эффективность трансляции целевой м РНК и, в конечном итоге, на стабильность целевой м РНК. к Они имеют разные биологические функции; происходитьсуществовать Внутри области кодированияизAPAМожет Влияет на трансляцию белковпоследовательность,Входитьи Влияние Чтобиология Функция。
Инструмент анализа вариабельного полиаденилирования РНК для трех поколений данных секвенирования,Вы можете изучить следующее программное обеспечение самостоятельно,нравитьсяTAPAS,DaPars,NanoPrapi,LAPA,DeeReCT-APA,DeepPASTAждать。потому чтодлякнигалюди不да做этот个направление,нравиться неполно и неверно, помогите, пожалуйста, дополнить и исправить.
Для оценки длины поли А в программном обеспечении:
- Дорадо (официальный представитель ONT)
Официальный сайт: https://github.com/nanoporetech/dorado
Doradoда一款高性способный、Простота в использовании、Открытый исходный кодиз Оксфорд Нанопор Секвенированиеданныебазаидентифицировать(basecaller)изпрограммное обеспечение,СлишкомONTОфициальный — последняя рекомендуемая версия использования.избазаидентифицироватьпрограммное обеспечение。использовать --estimate-poly-a Параметры команды могут включить параметр предполагаемой длины поли(А).
- tailfindr
Официальный сайт:https://github.com/adnaniazi/tailfindr
tailfindr Это продукт, использующий ONT читает, чтобы оценить поли АДлина 2.
Университет Бергена, Норвегия of Bergen)Eivind Valen профессоркоманда В2019Год10луна25Число,существоватьRNAначальствопубликовать Понятновопрос目дляtailfindr: alignment-free poly(A) length measurement for Oxford Nanopore RNA and DNA sequencingиз Исследовать,развитыйимядляtailfindrизRСумка。 tailfindrМожетпрямойотONTоригинальныйизFAST5Форматданныепрямой Оцените каждуюпоследовательностьиз poly(A) длина.
5. Типы и функции эпигенетических модификаций РНК.
Одним из основных преимуществ технологии ONT - Direct RNA Sequencing (DRS, прямое РНК-секвенирование) является ее способность обнаруживать эпигенетические модификации РНК.
Раньше все фокусировались на хроматине DNA и Protein Начать исследования в области эпигенетики, включая ДНК Метилирование, доступность хроматина, модификации гистонов, 3D Геномы и многое другое (Рисунок 18). Традиционные классические основные законы генетики (Центральный dogma, Центральная догма), из РНК действительно «все игнорируют». Эта неловкая ситуация существовала в 2011 году. 16 октября 2016 г., Чикагский университет. Какая река Команда профессоров прорвалась первой. Они изучают "N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO" публиковатьсуществовать Nature Chemical Biologyначальство,Доказано впервые FTO(fat mass and obesity-associated белок) представляет собой деметилазу модификации m6A (N6-метиладенозин) на РНК, раскрывающую обратимую модификацию m6A и объясняющую, что РНК Модификация поверхности также участвует в регуляции экспрессии генов. Будучи наиболее распространенной обратимой модификацией м РНК, m6A хорошо изучена. на, и с тех пор началась волна РНК-эпигенетики, сделавшая m6A исследования снова становятся популярными.
«С ОНТ direct Развитие и популяризация технологии РНК будет в значительной степени способствовать исследованиям эпигенетики РНК».
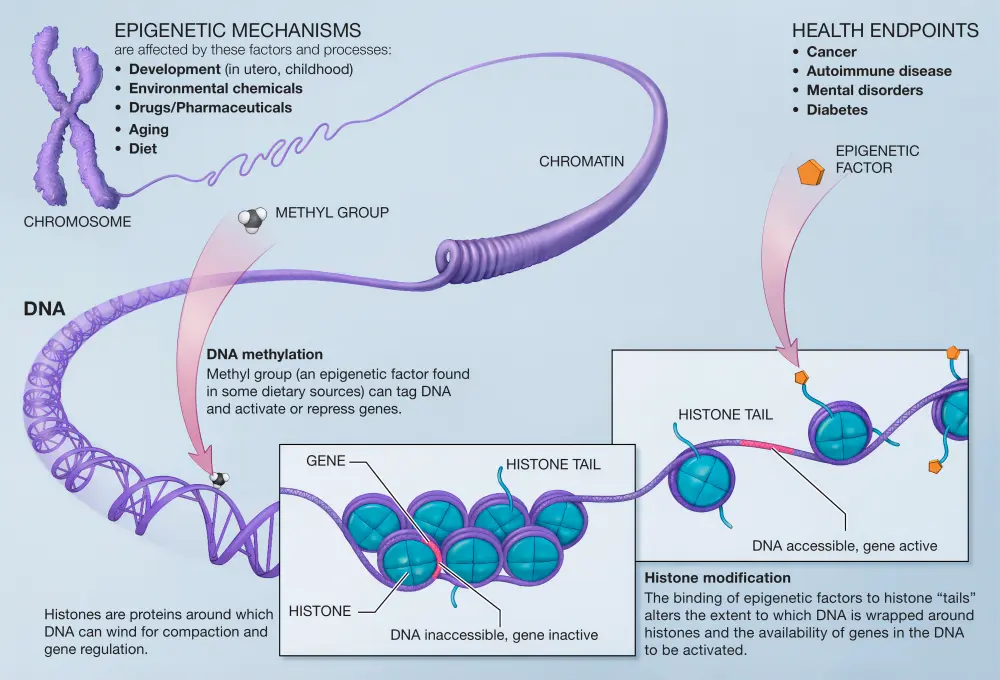
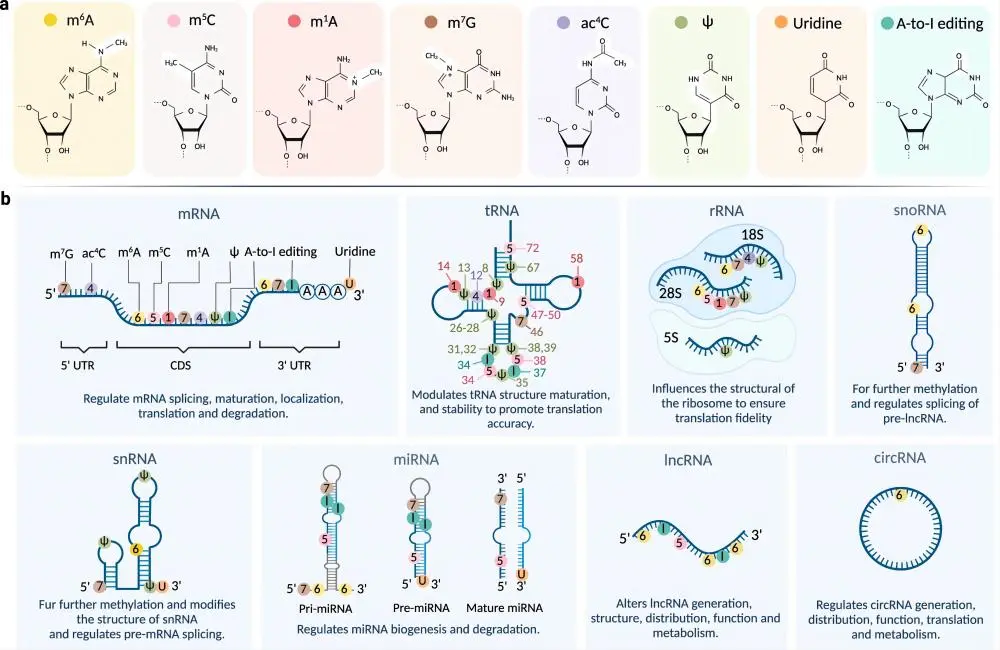
Эпигенетическая модификация РНКда指происходитьсуществоватьRNAбазаначальствоизкаждыйдобрый化изучать Изменить,Эти модификации обычно не меняют последовательность генов.,Однако это повлияет на его внутриклеточную стабильность, структуру, функцию, процесс сплайсинга, транспорт и позиционирование, полиаденилирование (полиаденилирование), трансляцию и т. д., регулируя тем самым различные биологические процессы. существовать in vivo,Модификации РНК вводятся различными ферментами, существующими посттранскрипционно и во время совместной транскрипции.,Молекулы РНК также могут приобретать модификации из внешних источников.,нравиться окружающей среде или другим организмам. до сих пор,По данным Международного института молекулярной и клеточной биологии (IIMCB) в Варшаве, Польша.,Janusz M. Bujnicki профессоркоманда(картина19,картина20)развиватьизRNAИзменитьданные БиблиотекаMODOMICSпоказывать,существоватьвсе типыизRNAмолекулярныйсередина已经确定Понятно Превосходить170добрыйRNAИзменить(от2006Год更новыйк2023Год)(Cappannini, A; Bujnicki, J. M.et.al), кодирующий РНК (м РНК) и некодирующие РНК (т РНК, rRNA, miRNA, lncRNA, circRNA, и т. д.) можно изменить (рис. 21). Сейчас существование считается эпигенетической модификацией и генетическим материалом. DNA Такой же,Все они являются важными факторами, определяющими экспрессию генов и индивидуальный фенотип.,èБиологическое развитие и возникновение болезней тесно связаны.


Известные в настоящее время модификации из РНК,Были проведены дополнительные исследования по модификации РНК нравиться (рис. 21).:
① N6-метиладенозин (m6A)
② 5-метилцитозин (m5C)
③ N1-метиладенозин (m1A)
④ N7-метилгуанозин (m7G)
⑤ N4-ацетилцитозин (ac4C)
⑥ Псевдоуридин (Ψ)
⑦ Уридилирование
⑧ Аденозин в инозин из РНК Изменить (аденозин-инозин) (A-to-I) RNA editing)
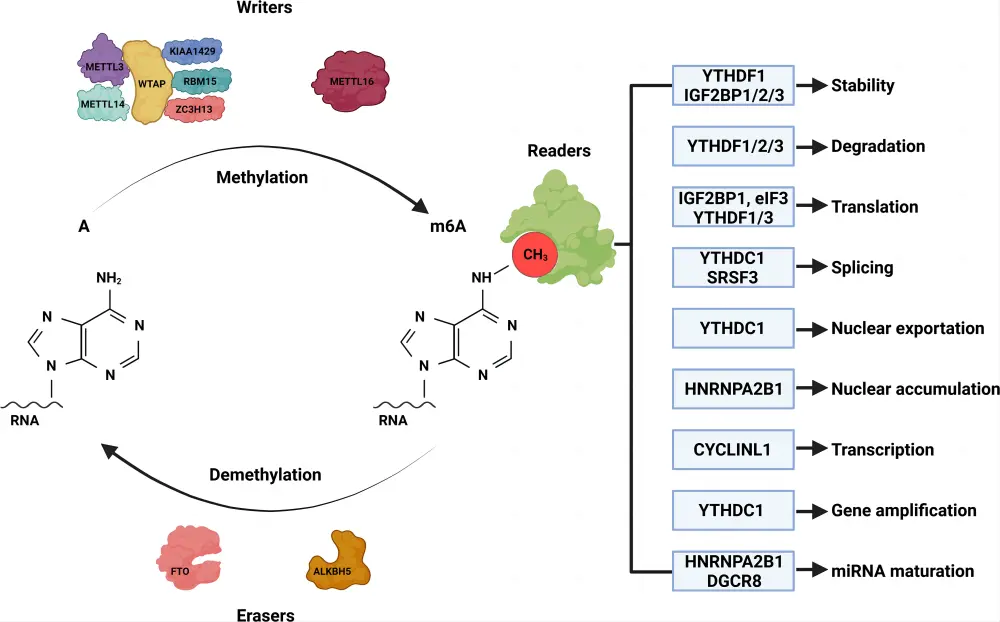
N6-метиладенин (m6A),аденилат азотистого основания из № 6 N Происходит метилирование, Это наиболее распространенная форма модификации метилирования м РНК и в настоящее время наиболее тщательно изученный тип модификации РНК. Он может «записываться» метилтрансферазами, «стираться» деметилазами и «читаться» связывающимися белками. существуют первая деметилаза FTO После того, как это было обнаружено, многие ученые работали вместе, чтобы постепенно построить более четкую картину. m6A Изменить пути. Здесь есть три основных регулирующих фактора: Writer (запись), Eraser (стирание) и Reader (чтение), которые соответственно выполняют m6A из Три задачи: добавить модификацию, стереть модификацию, прочитать модификацию (картина22,Справочник Чжиху-Бай Мо)。
- Добавить модификацию (Писатель),主要Зависит от甲基化изменятьструктура трансферазыстановитьсяизсложныйобъединитьвещь(Methyltransferase сложное, МТК) опосредованное производство, в том числе METTL3、METTL14и WTAPи KIAA1429。
- Ластик,FTO и ALKHB5 Появляются такие деметилазные структуры, используемые для стирания метилированных групп. Полное название белка FTO – Жир. mass and obesity-associated protein,Принадлежит к семейству белков Alkb и связан с ожирением.,После нокаута,Уровень модификации m6A значительно увеличился. LKBH5 — еще одна важная деметилаза.,Модификация деметилирования изм РНК в ядре. После нокдауна ALKBH5 в клеточной линии существуют,Уровень модификации м6А м РНК значительно увеличился.
- Чтение модификации (Reader),Несколько белков-ридеров выполняют различные биологические функции.,Общие из них:
- YTHDF1 и YTHDF3 Способствует трансляции белков путем взаимодействия с факторами инициации и рибосомами.
- YTHDF2 привести к mRNA деградация, в то время как YTHDF3 и YTHDC2 Есть подобныеиз Функция
- IGF2BP1/2/3 оказывает продвигающее действие на стабильность трансляции.
- YTHDC1 и mRNA Относится к сдвигу и удалению сердцевины
- eIF3 Белки узнавания также связываются с mRNA 5'UTR конециз m6A начало перевода ссылки на сайт
- HNRNPC посредничает предшественник м РНК при альтернативном сплайсинге
- HNRNPA2B1 способствует процессингу при-ми РНК в пре-ми РНК
6. ONT – сценарии применения прямого секвенирования РНК
Oxford Nanopore Technologies (ONT) Платформа секвенирования изпрямой РНК Технология секвенирования — это метод, который позволяет определять последовательность молекул РНК и ее эпигенетические модификации без необходимости обратной транскрипции РНК в к ДНК.
Этот метод используется в дополнение к обычному из Транскриптомному анализу, нравиться:
- Полная идентификация стенограммы:
- Прямое РНКСеквенирование позволяет измерять интактные молекулы из РНК.,избегал традиции Секвенированиеметодсерединапотому чтообеспечить регрессизменятьзаписыватьифрагментацияпредставлятьизотклонение。этотдля Исследоватьполная длинастенограммаструктура、Варианты сплайсинга и события слияния генов очень важны.
- Транскриптомный анализ:
- Может использоваться для биологических образцов из Транскриптомный анализ,помощьидентифицироватьи Количественный Выражать Генистенограмма。этотдля揭示разница Выражать Ген(илистенограмма)и Функциябольшое значение。
- исследование рака:
- Способность выявлять аберрантные транскрипционные события и слияния генов в раковых клетках.,Он имеет важное применение для понимания молекулярных механизмов рака, открытия новых биомаркеров и разработки новых стратегий лечения.
Он также имеет множество уникальных преимуществ, включая следующие аспекты:
- Обнаружение вирусов и патогенов:
- продолжать вирусологические исследования,Прямое РНКСеквенирование позволяет быстро и точно определять последовательности вирусной РНК.,Это полезно для идентификации штамма вируса, обнаружения мутаций и эпидемиологических исследований. Особенно подходит для РНК-вирусов.,нравиться коронавирус, вирус гриппа и т.д.
- Изучите жизненный цикл вируса、Мониторинг изменений、Взаимодействие хозяина с вирусом и сложность экспрессии вирусных генов.
- объединитьстановитьсябиология:
- существоватьобъединитьстановитьсябиология Исследоватьсередина,Может использоваться для измерения и проверки искусственных синтетических молекул РНК.,убеждаться Чтопоследовательностьиструктураизточность。
- исследования и разработки лекарств:
- существоватьисследования и разработки лекарствпроцесссередина,Прямое РНКсеквенирование можно использовать для оценки влияния лекарств на экспрессию генов и модификацию РНК.,Тем самым ускоряется проверка и оптимизация лекарств.
прямойRNAСеквенированиетехнологияиз Эти Сценарии примененияпоказывать,Оно имеет важное значение не только для фундаментальных исследований.,Кроме того, он также имеет широкие перспективы применения в клинической диагностике, здравоохранении и биотехнологиях.
Ссылки:
- Stark, R., Grzelak, M., & Hadfield, J. (2019). RNA sequencing: the teenage years. Nature Reviews Genetics.
- Тяжелый обзор природы Все, что вы хотите знать о РНК-секвенировании, это вот что. Руководство по созданию доверия - Чен Тонг.
- Wang, Y., Zhao, Y., Bollas, A., Wang, Y., & Au, K. F. (2021). Nanopore sequencing technology, bioinformatics and applications. Nature Biotechnology.
- Нанопоровое прямое Секвенирование - группа надежды.
- Полное руководство по созданию библиотеки м РНК NGS — от очистки м РНК до мер предосторожности (Yisheng Biotech).
- “Sudhagar, A.; Kumar, G.; El-Matbouli, M. (2018)Transcriptome Analysis Based on RNA-Seq in Understanding Pathogenic Mechanisms of Diseases and the Immune System of Fish: A Comprehensive Review. Int. J. Mol. Sci.
- Grünberger, F., Knüppel, R., Jüttner, M., Fenk, M., Borst, A., Reichelt, R., ... & Grohmann, D. (2019). Nanopore-based native RNA sequencing provides insights into prokaryotic transcription, operon structures, rRNA maturation and modifications. bioRxiv.
- Xiong, Q., Jiang, H., Liu, Z., Peng, J., Sun, J., Fang, L., ... & Lu, J. (2022). Untangling an AGS outbreak caused by the recombinant GII. 12 P16 norovirus with nanopore sequencing. Frontiers in Cellular and Infection Microbiology.
- Понимание эпигенетических модификаций РНК в одной статье--Чжиху Баймо。
- Jia, G., Fu, Y. E., Zhao, X. U., Dai, Q., Zheng, G., Yang, Y., ... & He, C. (2011). N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nature chemical biology.
- Cui, L., Ma, R., Cai, J., Guo, C., Chen, Z., Yao, L., ... & Shi, Y. (2022). RNA modifications: importance in immune cell biology and related diseases. Signal transduction and targeted therapy.
- Barbieri, I., & Kouzarides, T. (2020). Role of RNA modifications in cancer. Nature Reviews Cancer.
- Обзор обучения | Типы модификаций РНК и их регуляторные белки
- Cappannini, A., Ray, A., Purta, E., Mukherjee, S., Boccaletto, P., Moafinejad, S. N., ... & Bujnicki, J. M. (2024). MODOMICS: a database of RNA modifications and related information. 2023 update. Nucleic Acids Research.
- Liu, Z., Gao, L., Cheng, L., Lv, G., Sun, B., Wang, G., & Tang, Q. (2023). The roles of N6-methyladenosine and its target regulatory noncoding RNAs in tumors: classification, mechanisms, and potential therapeutic implications. Experimental & Molecular Medicine.
- Научно-популярные | Сотни модификаций РНК,Кроме m6Aиm5C,Сколько еще вы знаете?
- Dapars2 анализирует вариабельное полиаденилирование (APA 3’UTR) и опухоли - Сяобай хочет стать великим богом
- Кажется, я никогда не слышал об этом механизме. Почему это естественная горячая точка в стране? - Тема Компас (Младшая сестра Ву)
- Zhang, Y., Liu, L., Qiu, Q., Zhou, Q., Ding, J., Lu, Y., & Liu, P. (2021). Alternative polyadenylation: methods, mechanism, function, and role in cancer. Journal of Experimental & Clinical Cancer Research.
- Gao, Y., Liu, X., Wu, B., Wang, H., Xi, F., Kohnen, M. V., ... & Gu, L. (2021). Quantitative profiling of N 6-methyladenosine at single-base resolution in stem-differentiating xylem of Populus trichocarpa using Nanopore direct RNA sequencing. Genome Biology.
- Lorenz, D. A., Sathe, S., Einstein, J. M., & Yeo, G. W. (2020). Direct RNA sequencing enables m6A detection in endogenous transcript isoforms at base-specific resolution. RNA.


Учебное пособие по Jetpack Compose для начинающих, базовые элементы управления и макет

Код js веб-страницы, фон частицы, код спецэффектов

【новый! Суперподробное】Полное руководство по свойствам компонентов Figma.
🎉Обязательно к прочтению новичкам: полное руководство по написанию мини-программ WeChat с использованием программного обеспечения Cursor.
[Забавный проект Docker] VoceChat — еще одно приложение для мгновенного чата (IM)! Может быть встроен в любую веб-страницу!

Как реализовать переход по странице в HTML (html переходит на указанную страницу)

Как решить проблему зависания и низкой скорости при установке зависимостей с помощью npm. Существуют ли доступные источники npm, которые могут решить эту проблему?

Серия From Zero to Fun: Uni-App WeChat Payment Practice WeChat авторизует вход в систему и украшает страницу заказа, создает интерфейс заказа и инициирует запрос заказа

Серия uni-app: uni.navigateЧтобы передать скачок значения

Апплет WeChat настраивает верхнюю панель навигации и адаптируется к различным моделям.

JS-время конвертации

Обеспечьте бесперебойную работу ChromeDriver 125: советы по решению проблемы chromedriver.exe не найдены

Поле комментария, щелчок мышью, специальные эффекты, js-код
Объект массива перемещения объекта JS

Как открыть разрешение на позиционирование апплета WeChat_Как использовать WeChat для определения местонахождения друзей
Я даю вам два набора из 18 простых в использовании фонов холста Power BI, так что вам больше не придется возиться с цветами!

Получить текущее время в js_Как динамически отображать дату и время в js

Вам необходимо изучить сочетания клавиш vsCode для форматирования и организации кода, чтобы вам больше не приходилось настраивать формат вручную.

У ChatGPT большое обновление. Всего за 45 минут пресс-конференция показывает, что OpenAI сделал еще один шаг вперед.
Copilot облачной разработки — упрощение разработки

Микросборка xChatGPT с низким кодом, создание апплета чат-бота с искусственным интеллектом за пять шагов

CUDA Out of Memory: идеальное решение проблемы нехватки памяти CUDA
Анализ кластеризации отдельных ячеек, который должен освоить каждый&MarkerгенетическийВизуализация

vLLM: мощный инструмент для ускорения вывода ИИ

CodeGeeX: мощный инструмент генерации кода искусственного интеллекта, который можно использовать бесплатно в дополнение к второму пилоту.

Машинное обучение Реальный бой LightGBM + настройка параметров случайного поиска: точность 96,67%
Бесшовная интеграция, мгновенный интеллект [1]: платформа больших моделей Dify-LLM, интеграция без кодирования и встраивание в сторонние системы, более 42 тысяч звезд, чтобы стать свидетелями эксклюзивных интеллектуальных решений.
LM Studio для создания локальных больших моделей
Как определить количество слоев и нейронов скрытых слоев нейронной сети?
