Полноразмерный транскриптом | Конвейер анализа данных секвенирования третьего поколения Iso-Seq (PacBio) (3) — SQANTI3 v5.2
Makeuse Секвенирование длиной в три поколения выполнено полностью транскриптом Высокопроизводительное секвенирование открывает путь к открытию тысяч новых транскриптов, даже у хорошо аннотированных видов млекопитающих, оно также предоставляет мощные технические средства для углубленной характеристики изменений генов на уровне транскриптов;
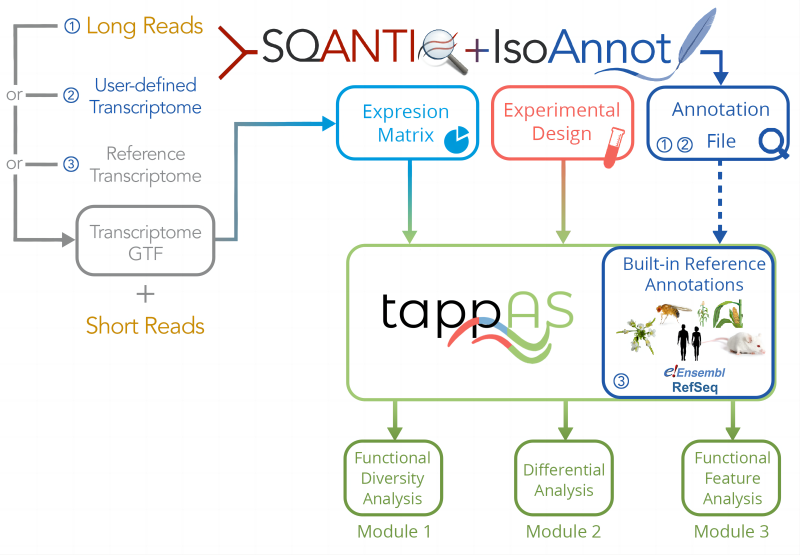
Functional IsoTranscriptomics (FIT) Университет Флориды of Florida)Ana Conesa Команда профессоров (Геномика of Gene Expression Lab, Конвейер для биоинформатического анализа на уровне изоформ транскриптов, разработанный ConesaLab с целью предоставить комплексное решение для полноразмерных транскриптомов. (картина1)。SQANTI 3 Являясь первым модулем конвейера FIT, он предназначен для обеспечения контроля качества и фильтрации транскриптомов длительного считывания, которые часто содержат артефакты и ложноположительные результаты. Следовательно, для полного транскриптом Калибровка выполняетсяFITПредварительные условия для анализа,и производить надежные、Биологически обоснованный вывод/Предположения имеют решающее значение。SQANTI 3 Это СКАНТИ Последняя версия инструмента (выпуск), который объединяет SQANT 1 и SQANTI 2 функции и добавлены новые функции , лучшая глубокая характеристика полноразмерных транскриптов 。
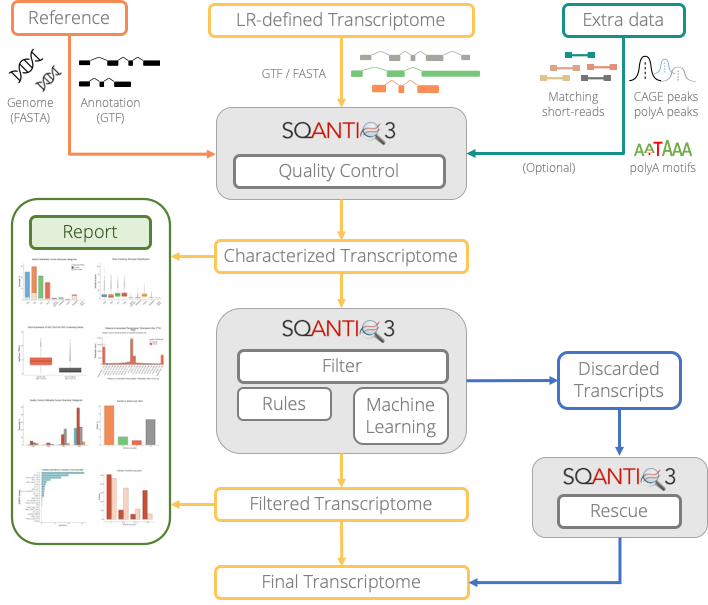
SQANTI 3 Предназначен в первую очередь для выполнения двух одинаково важных задач. (Рисунок 2):
1.Текущая классификация транскриптома и контроль качества длинных считываемых последовательностей (SQANTI3 QC)。SQANTI 3 определенный isoform Справочные аннотированные категории и подкатегории в сочетании с рядом атрибутов и описаний на уровне транскриптов позволяют пользователям тщательно изучать свойства своих моделей изоформ и выявлять потенциальные проблемы, которые возникают во время подготовки библиотеки и обработки необработанных данных.
2.Длинночитаемые последовательности определяют транскриптом artifacts Фильтр (SQANTI3 filter)。выгодаиспользовать SQANTI 3 Вычислив это, пользователи могут удалить потенциальные ложноположительные изоформы из своих транскриптомов. Это особенно важно, учитывая предвзятость и подводные камни современных экспериментальных рабочих процессов секвенирования с длительным чтением.
Чтобы получить более глубокое понимание этих двух шагов,Мы поощряем чтениеСКАНТИ оригинальный текст。Однако,Недавно мы былиSQANTI 3 В рабочий процесс добавлен последний шаг (SQANTI3 rescue):
3.Спасение эталонной расшифровки (спасение SQANTI3)。Стремление удалить аннотацию генома по ссылкеartifacts。Этот модуль предназначен для сохранения разнообразия транскриптомов.,Это делается для того, чтобы не потерять тех, кто пользуется доверием.junction chainsСтенограмма。запустивSQANTI3 rescueпрограмма,SQANTI 3 Эталонные транскрипты, которым доверяют как соответствующие удаленным артефактам, будут выбраны и добавлены обратно в отфильтрованный транскриптом.
использовать SQANTI 3 После создания высококачественного транскриптома последующие этапы включают в себя (рис. 1):
- Функциональная аннотация модели изоформы,Включает в себя текущие функциональные возможности местоположения,нравитьсяmotifs、Структурные области и т. д.。
IsoAnnotэтоиспользовать Направоisoformруководитьde novoИнструменты аннотаций。В настоящее время в разработке,Но использовать домохозяйства могут SQANTI 3 Запуск внутри или снаружиIsoAnnotLite,Выведите функциональные особенности из других аннотированных транскриптомов. - делатьиспользовать
tappASруководить Основанное на выраженияхиз Функциональный анализ。tappASэтоJava GUIотвечатьиспользоватьпрограмма,Его преимущества: использование выражений и информации о аннотациях домена и мотивов.,Чтобы получить представление о функциональном влиянии экспрессии альтернативно сплайсированных изоформ.
Перед запуском SQANTI 3: Рекомендуемый процесс обработки последовательности длинного чтения:
Вот наш рекомендуемый рабочий процесс, включая создание SQANTI 3 Как лучше вводить файлы и что делать после проверки качества и фильтрации:
- Объединение образцов:Хотя мы знаем некоторыеиспользовать Пользователи могут повторять эксперименты из несколькихи/Или данные длинной считываемой последовательности были получены из образца,Но мы рекомендуем объединить все данные выборки длительного чтения.,построить единый транскриптом для каждого эксперимента.
- Используйте предпочитаемый вами инструмент построения транскриптома для обработки данных длительного чтения.。Мы не рекомендуем использовать необработанные лонгриды.(raw long reads)начальстводелатьиспользовать SQANTI 3,Потому что он не предназначен для контроля качества данных длинных считываний последовательностей.
- Объединение моделей транскриптов。в целом,Конвейеры обработки последовательностей длительного чтения генерируют большое количество крайне избыточныхisoformМодель。ямы предлагаемсуществоватьделатьиспользоватьSQANTI 3 вперед,Первыйиспользовать
cDNA_Cupcake(сейчас этоisoseq collapse)илиTAMA CollapseПодождите, пока инструменты объединят лишниеisoform,Обеспечить количество и качество изоформ. - Контроль качества и фильтрация:Мы настоятельно рекомендуемиспользоватьпользователи как можно тщательнее изучают свои длинные считываемые транскриптомы с определенной последовательностью,Включает скрининг транскриптома для удаления возможных ложноположительных изоформ.,Это часто встречается в транскриптомах, полученных из последовательностей длительного чтения.
- Используйте короткое/длинное чтение и соответствующие инструменты для количественной оценки отфильтрованного транскриптома.。Мы не рекомендуем преобразовывать входные данные в
SQANTI 3Оценка уровня экспрессиииспользоватьв дальнейшем анализе:Это толькоиспользоватьв целях контроля качества。После того, как все они будут удалены из транскриптомаartifacts,Эти последовательности можно сравнить для получения более точной количественной оценки.
1. Установка SQANTI3 v5.2
- Загрузка SQANTI3
$ wget https://github.com/ConesaLab/SQANTI3/archive/refs/tags/v5.2.tar.gz
$ tar -xvf v5.2.tar.gz- СКАНТИ3
condaСоздано средой
$ cd SQANTI3
$ conda env create -f SQANTI3.conda_env.yml
$ source activate SQANTI3.env- Установить
gtfToGenePred, добавьте запущенную программу в путь
#Воля gtfToGenePred присоединиться PATH, измените его в соответствии со своим путем.
$ echo PATH=/mnt/data/home/mli/Desktop/Software/SQANTI3-5.2/utilities:$PATH >> ~/.bashrc && source ~/.bashrc
#Воля SQANTI3 pythonСкриптприсоединиться PATH, измените его в соответствии со своим путем.
$ echo PATH=/mnt/data/home/mli/Desktop/Software/SQANTI3-5.2:$PATH >> ~/.bashrc && source ~/.bashrc- Установить
cDNA_Cupcakeпрограммное обеспечение
#голова Первый Активировать SQANTI3
$ source activate SQANTI3.env
#Затем скачайте cDNA_Cupcake
(SQANTI3.env)$ git clone https://github.com/Magdoll/cDNA_Cupcake.git
(SQANTI3.env)$ cd cDNA_Cupcake
#УстановитьcDNA_Cupcake
(SQANTI3.env)$ python setup.py build
(SQANTI3.env)$ python setup.py install2. Запустите SQANTI3
1. Активировать SQANTI3 conda среда
(base)-bash-4.1$ conda activate SQANTI3.env
(SQANTI3.env)-bash-4.1$2. Пучок cDNA_Cupcake/sequence папкапутьприсоединиться$PYTHONPATH
#официальный учебник
(SQANTI3.env)-bash-4.1$ export PYTHONPATH=$PYTHONPATH:<path_to>/cDNA_Cupcake/sequence/
(SQANTI3.env)-bash-4.1$ export PYTHONPATH=$PYTHONPATH:<path_to>/cDNA_Cupcake/
#Фактическая операция
(SQANTI3.env)-bash-4.1$ export PYTHONPATH=$PYTHONPATH:/mnt/data/home/mli/Desktop/Software/cDNA_Cupcake-master/sequence/
(SQANTI3.env)-bash-4.1$ export PYTHONPATH=$PYTHONPATH:/mnt/data/home/mli/Desktop/Software/cDNA_Cupcake-master3. Запустите SQANTI3 QC sqanti3_qc.py
- Официальный пример
#Входить SQANTI3-5.2 папка
#Исправлять UHR_chr22_short_reads.fofn Путь к файлу следующий:
example/UHR_Rep1_chr22.R1.fastq.gz example/UHR_Rep1_chr22.R2.fastq.gz
example/UHR_Rep2_chr22.R1.fastq.gz example/UHR_Rep2_chr22.R2.fastq.gz
#бегать
$ sqanti3_qc.py example/UHR_chr22.gtf example/gencode.v38.basic_chr22.gtf example/GRCh38.p13_chr22.fasta \
--CAGE_peak data/ref_TSS_annotation/human.refTSS_v3.1.hg38.bed \
--polyA_motif_list data/polyA_motifs/mouse_and_human.polyA_motif.txt \
-o UHR_chr22 -d example/SQANTI3_output -fl example/UHR_abundance.tsv \
--short_reads example/UHR_chr22_short_reads.fofn --cpus 4 --report both 
- Реальный случай, на примере первой части данных URHH:
$ python sqanti3_qc.py example/UHRR.collapsed.gff example/GRCh38.gtf example/GRCh38.fa \
-o UHRR -d example/SQANTI3_UHRR_Output -fl example/UHRR.collapsed.flnc_count.txt \
--isoAnnotLite --gff3 example/human_tappas_gencode_annotation_file.gff3 \
--cpus 20 --report both
специфическийИнтерпретация файлов результатовВы можете обратиться к официальной документации:Understanding-the-output-of-SQANTI3-QC 。
Советы:
1). Когда я запускаю другую папку,Продолжайте сообщать об ошибках,После различных тестов входных файлов,Я думаю, это проблема с зависимостью файлов,Поскольку пример выполняется успешно;И вSQANTI3-5.2документвнутритранспорт ХОРОШО就可以正常транспорт ХОРОШО,Просто нужно Пучок Файлы, включая эталонный геном и файлы его аннотаций, копируются вexapmleпапкавнутри。
2). isoseq collapse произведено *.collapsed.gffда.gff2Формат,иsqanti3_qc.pyвходной файл.gtfФормат Такой же,такда Проходитьиспользоватьиз。ненужныйиспользоватьgffreadруководить Конвертировать。
3).gffreadиз Установитьиделатьиспользовать。
$ conda install -c bioconda gffread
#GFFconvertGTF
$ gffread input.gff3 -T -o out.gtf
#GTFconvertGFF3
$ gffread input.gtf -o out.gff34).cDNA_cupcake Обновления остановлены,официальнымiso-seqпоглощено и заменено,поэтомуiso-seq collapse иTAMA collapseизвыход.gffдокумент都официальным Рекомендуется какsqanti3_qc.pyизвходной файл。
4. SQANTI3 QC Параметры
usage: sqanti3_qc.py [-h] [--min_ref_len MIN_REF_LEN] [--force_id_ignore]
[--aligner_choice {minimap2,deSALT,gmap,uLTRA}]
[--CAGE_peak CAGE_PEAK] [--polyA_motif_list POLYA_MOTIF_LIST]
[--polyA_peak POLYA_PEAK] [--phyloP_bed PHYLOP_BED]
[--skipORF] [--is_fusion] [--orf_input ORF_INPUT]
[--fasta] [-e EXPRESSION] [-x GMAP_INDEX]
[-t CPUS] [-n CHUNKS] [-o OUTPUT] [-d DIR]
[-c COVERAGE] [-s SITES] [-w WINDOW] [--genename]
[-fl FL_COUNT] [-v] [--saturation]
[--report {html,pdf,both,skip}]
[--isoAnnotLite] [--gff3 GFF3]
[--short_reads SHORT_READS] [--SR_bam SR_BAM]
[--isoform_hits]
[--ratio_TSS_metric {max,mean,median,3quartile}]
isoforms annotation genome
Structural and Quality Annotation of Novel Transcript Isoforms
positional arguments:
isoforms Isoforms (FASTA/FASTQ) or GTF format. It is recommended to
provide them in GTF format, but if it is needed to map the sequences
to the genome use a FASTA/FASTQ file with the
--fasta option.
annotation Reference annotation file (GTF format)
genome Reference genome (Fasta format)
options:
-h, --help show this help message and exit
--min_ref_len MIN_REF_LEN
Minimum reference transcript length (default: 200 bp)
--force_id_ignore Allow the usage of transcript IDs non related with
PacBio's nomenclature (PB.X.Y)
--aligner_choice {minimap2,deSALT,gmap,uLTRA}
--CAGE_peak CAGE_PEAK
FANTOM5 Cage Peak (BED format, optional)
--polyA_motif_list POLYA_MOTIF_LIST
Ranked list of polyA motifs (text, optional)
--polyA_peak POLYA_PEAK
PolyA Peak (BED format, optional)
--phyloP_bed PHYLOP_BED
PhyloP BED for conservation score (BED, optional)
--skipORF Skip ORF prediction (to save time)
--is_fusion Input are fusion isoforms, must supply GTF as input
--orf_input ORF_INPUT
Input fasta to run ORF on. By default, ORF is run on genome-corrected
fasta - this overrides it. If input is fusion (--is_fusion), this must be provided for ORF prediction.
--fasta Use when running SQANTI by using as input a FASTA/FASTQ with the sequences of isoforms
-e EXPRESSION, --expression EXPRESSION
Expression matrix (supported: Kallisto tsv)
-x GMAP_INDEX, --gmap_index GMAP_INDEX
Path and prefix of the reference index created by gmap_build. Mandatory
if using GMAP unless -g option is specified.
-t CPUS, --cpus CPUS Number of threads used during alignment by aligners. (default: 10)
-n CHUNKS, --chunks CHUNKS
Number of chunks to split SQANTI3 analysis in for speed up (default: 1).
-o OUTPUT, --output OUTPUT
Prefix for output files.
-d DIR, --dir DIR Directory for output files. Default: Directory where the script was run.
-c COVERAGE, --coverage COVERAGE
Junction coverage files (provide a single file, comma-delmited filenames,
or a file pattern, ex: "mydir/*.junctions").
-s SITES, --sites SITES
Set of splice sites to be considered as canonical (comma-separated list of splice
sites). Default: GTAG,GCAG,ATAC.
-w WINDOW, --window WINDOW
Size of the window in the genomic DNA screened for Adenine content downstream of TTS
--genename Use gene_name tag from GTF to define genes. Default: gene_id used to define genes
-fl FL_COUNT, --fl_count FL_COUNT
Full-length PacBio abundance file
-v, --version Display program version number.
--saturation Include saturation curves into report
--report {html,pdf,both,skip}
select report format --html --pdf --both --skip
--isoAnnotLite Run isoAnnot Lite to output a tappAS-compatible gff3 file
--gff3 GFF3 Precomputed tappAS species specific GFF3 file. It will serve as reference
to transfer functional attributes
--short_reads SHORT_READS
File Of File Names (fofn, space separated) with paths to FASTA or FASTQ from
Short-Read RNA-Seq. If expression or
coverage files are not provided, Kallisto (just for pair-end
data) and STAR, respectively, will be run to calculate them.
--SR_bam SR_BAM Directory or fofn file with the sorted bam files of Short Reads RNA-Seq mapped
against the genome
--isoform_hits Report all FSM/ISM isoform hits in a separate file
--ratio_TSS_metric {max,mean,median,3quartile}
Define which statistic metric should be reported in the ratio_TSS column5. Запустите SQANTI3 FILTER sqanti3_filter.py
- Запуск фильтра правил
#учебник
$ python sqanti3_filter.py rules path/to/classification.txt
#Фактическая операция
$ python sqanti3_filter.py rules example/SQANTI3_UHRR_Output/UHRR_classification.txt - Запуск фильтра машинного обучения
#учебник
$ python sqanti3_filter.py ml path/to/classification.txt
#Фактическая операция
$ python sqanti3_filter.py ml example/SQANTI3_UHRR_Output/UHRR_classification.txt 6. Запустите SQANTI3 RESCUEsqanti3_rescue.py
#rulesна самом деле выполняется
$ python sqanti3_rescue.py rules --isoforms *_corrected.fasta --gtf *.filtered.gtf -f GRCh38.gtf -k UHRR_classification.txt
#машинное обучение в действии
$ python sqanti3_rescue.py ml --isoforms *_corrected.fasta --gtf *.filtered.gtf -f GRCh38.gtf -k UHRR_classification.txt3. Проблемы, с которыми вы можете столкнуться при установке SQANTI3.
1. Если вы столкнулись со следующей проблемой:
Error compiling Cython file:
------------------------------------------------------------
...
exon_tree.insert_interval(Interval(e_start+offset, i+offset, index))
index += 1
tag = False
elif baseC[i] > 0 and (altC_pos[i] > threshSplit or altC_neg[i+1] < -threshSplit): # alt. junction found!
# end the current exon at i and start a new one at i + 1
print "alt. junction found at", i
^
------------------------------------------------------------
cupcake/tofu/branch/c_branch.pyx:30:22: Syntax error in simple statement list
Traceback (most recent call last):
File "/mnt/data/home/mli/Desktop/Software/cDNA_Cupcake-master/setup.py", line 25, in <module>
ext_modules = cythonize(ext_modules),
File "/mnt/data/home/mli/miniforge-pypy3/envs/SQANTI3.env/lib/python3.10/site-packages/Cython/Build/Dependencies.py", line 1154, in cythonize
cythonize_one(*args)
File "/mnt/data/home/mli/miniforge-pypy3/envs/SQANTI3.env/lib/python3.10/site-packages/Cython/Build/Dependencies.py", line 1321, in cythonize_one
raise CompileError(None, pyx_file)
Cython.Compiler.Errors.CompileError: cupcake/tofu/branch/c_branch.pyxРешение:
редактировать в файле setup.py Строка 25:
Пучок ext_modules = cythonize(ext_modules),
Изменить на ext_modules = cythonize(ext_modules, language_level = "2"),
Запустите еще раз:
$ python setup.py build2. Если вы столкнулись со следующей проблемой:
SQANTI3.env) mli@ca496d1fda31:~$ sqanti3_qc.py
Traceback (most recent call last):
File "/mnt/data/home/mli/Desktop/Software/SQANTI3-5.2/sqanti3_qc.py", line 20, in <module>
from scipy import mean
ImportError: cannot import name 'mean' from 'scipy' (/mnt/data/home/mli/miniforge-pypy3/envs/SQANTI3.env/lib/python3.10/site-packages/scipy/__init__.py)Решение:
проверено,отвечать ДолжендаpythonВерсияизвопрос。SQANTI3.cond_env.ymlНет.29ХОРОШО,- python>=3.7.6,такcondaавтоматический Установитьдо настоящего времениизpython3.10。существоватьpythonсреда中транспорт ХОРОШОfrom scipy import meanСообщить об ошибке。я Пучок- python>=3.7.6Изменить на-python = 3.8.13。
- Удалите старую среду SQANTI3:
$ conda env remove --name SQANTI3.env- Повтор:
$ conda env create -f SQANTI3.conda_env.ymlТогда вот оно, ура!


Учебное пособие по Jetpack Compose для начинающих, базовые элементы управления и макет

Код js веб-страницы, фон частицы, код спецэффектов

【новый! Суперподробное】Полное руководство по свойствам компонентов Figma.
🎉Обязательно к прочтению новичкам: полное руководство по написанию мини-программ WeChat с использованием программного обеспечения Cursor.
[Забавный проект Docker] VoceChat — еще одно приложение для мгновенного чата (IM)! Может быть встроен в любую веб-страницу!

Как реализовать переход по странице в HTML (html переходит на указанную страницу)

Как решить проблему зависания и низкой скорости при установке зависимостей с помощью npm. Существуют ли доступные источники npm, которые могут решить эту проблему?

Серия From Zero to Fun: Uni-App WeChat Payment Practice WeChat авторизует вход в систему и украшает страницу заказа, создает интерфейс заказа и инициирует запрос заказа

Серия uni-app: uni.navigateЧтобы передать скачок значения

Апплет WeChat настраивает верхнюю панель навигации и адаптируется к различным моделям.

JS-время конвертации

Обеспечьте бесперебойную работу ChromeDriver 125: советы по решению проблемы chromedriver.exe не найдены

Поле комментария, щелчок мышью, специальные эффекты, js-код
Объект массива перемещения объекта JS

Как открыть разрешение на позиционирование апплета WeChat_Как использовать WeChat для определения местонахождения друзей
Я даю вам два набора из 18 простых в использовании фонов холста Power BI, так что вам больше не придется возиться с цветами!

Получить текущее время в js_Как динамически отображать дату и время в js

Вам необходимо изучить сочетания клавиш vsCode для форматирования и организации кода, чтобы вам больше не приходилось настраивать формат вручную.

У ChatGPT большое обновление. Всего за 45 минут пресс-конференция показывает, что OpenAI сделал еще один шаг вперед.
Copilot облачной разработки — упрощение разработки

Микросборка xChatGPT с низким кодом, создание апплета чат-бота с искусственным интеллектом за пять шагов

CUDA Out of Memory: идеальное решение проблемы нехватки памяти CUDA
Анализ кластеризации отдельных ячеек, который должен освоить каждый&MarkerгенетическийВизуализация

vLLM: мощный инструмент для ускорения вывода ИИ

CodeGeeX: мощный инструмент генерации кода искусственного интеллекта, который можно использовать бесплатно в дополнение к второму пилоту.

Машинное обучение Реальный бой LightGBM + настройка параметров случайного поиска: точность 96,67%
Бесшовная интеграция, мгновенный интеллект [1]: платформа больших моделей Dify-LLM, интеграция без кодирования и встраивание в сторонние системы, более 42 тысяч звезд, чтобы стать свидетелями эксклюзивных интеллектуальных решений.
LM Studio для создания локальных больших моделей
Как определить количество слоев и нейронов скрытых слоев нейронной сети?
