Полноразмерный микробный анализ 16S | PacBio Hifi Reads
Новое поколение микробных исследований – третье поколение полноразмерных 16S (Full-length 16S)
Сегодня исследования микробного сообщества полностью перешли на стадию анализа секвенирования, а нынешнее основное направление исследований находится в переходном периоде между ампликонами второго поколения и ампликонами третьего поколения. Анализ состава бактериального разнообразия на основе секвенирования третьего поколения может значительно повысить точность и полноту классификации и идентификации видов, а также более точно восстановить состав микробных сообществ в образцах. Одновременно с достижением обнаружения «высокого разрешения» он также обеспечивает. будущее Это заложило основу для углубленного изучения метаболических функций бактериальной флоры.
16S рибосомальная РНК (16S рибосомальная РНК), называемая 16S п РНК, является компонентом 30S рибосомальной субъединицы прокариот. Ген 16С р РНК присутствует во всех геномах бактерий.,Длина ок.1542 б.п., в том числе 10 Заповедная территория регион) и 9 Переменная площадь (Переменная регион), консервативный регион отражает генетическое родство между видами, а вариабельный регион отражает различия между видами. (Рисунок 1). 16S Ген р РНК с его умеренным молекулярным размером и низкой частотой мутаций является наиболее полезным и часто используемым молекулярным маркером в исследованиях по классификации бактерий. Обнаружение 16S посредством высокопроизводительного секвенирования ампликонов 16S Вариация последовательности и численность вариабельных областей р ДНК могут предоставить информацию о разнообразии и численности микробных сообществ в образцах и играть важную роль в классификации и идентификации микробов, микроэкологических исследованиях и т. д.
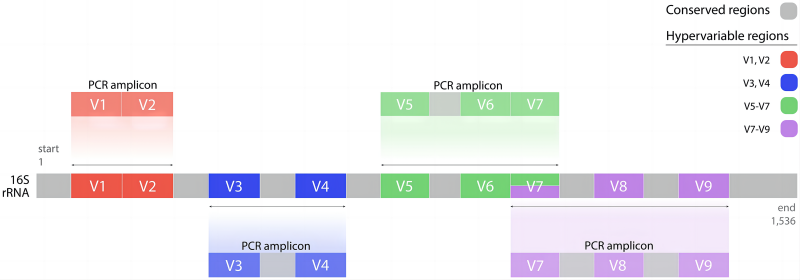
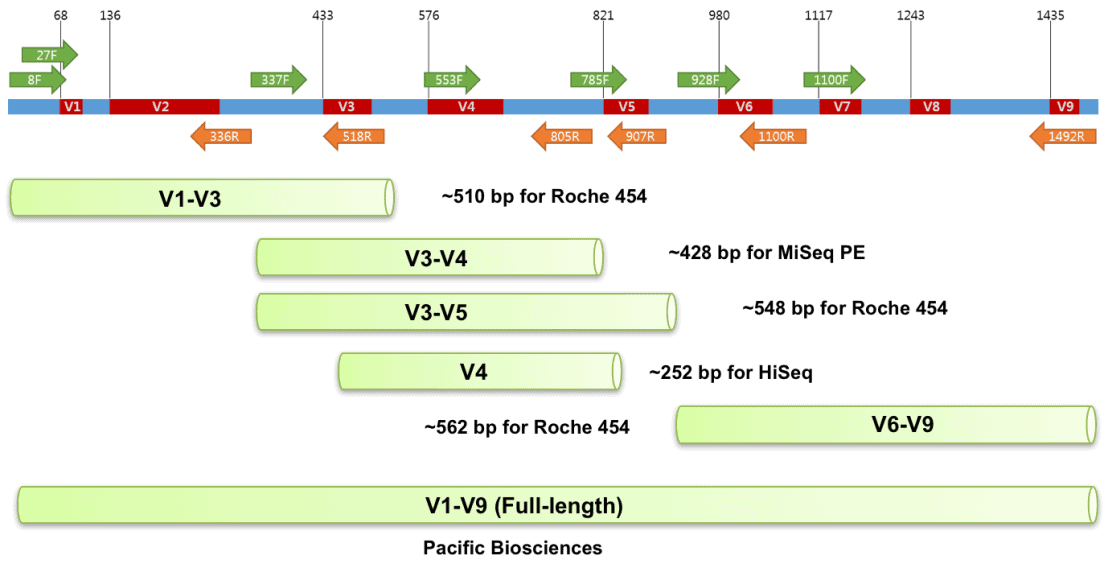
В 1990 году ученые впервые обнаружили последовательность 16S р РНК, присутствующую в образцах окружающей среды (1), и раскрыли ее исследовательский потенциал. С тех пор началась великолепная эра исследований микробного сообщества. Секвенирование 16S второго поколения имеет ограниченную длину амплифицированного фрагмента, составляющую всего 500-600 п.н. (двустороннее перекрытие), поэтому подбор вариабельных областей для ампликонов второго поколения представляет собой большую проблему. Отбор означает компромисс и потерю информации, как показано на рисунке. статью (3), с высокой долей неопознанных видов на уровне рода и вида (рис. 2). Секвенирование ампликона 16S третьего поколения использует праймеры 27F и 1492R для амплификации полноразмерного фрагмента (охватывающего область V1-V9), который может легко покрыть общую длину 16S около 1500 пар оснований и в общей сложности 9 вариабельных областей, что максимально увеличивает возможность разделения видов. идентификация (рисунок 3).
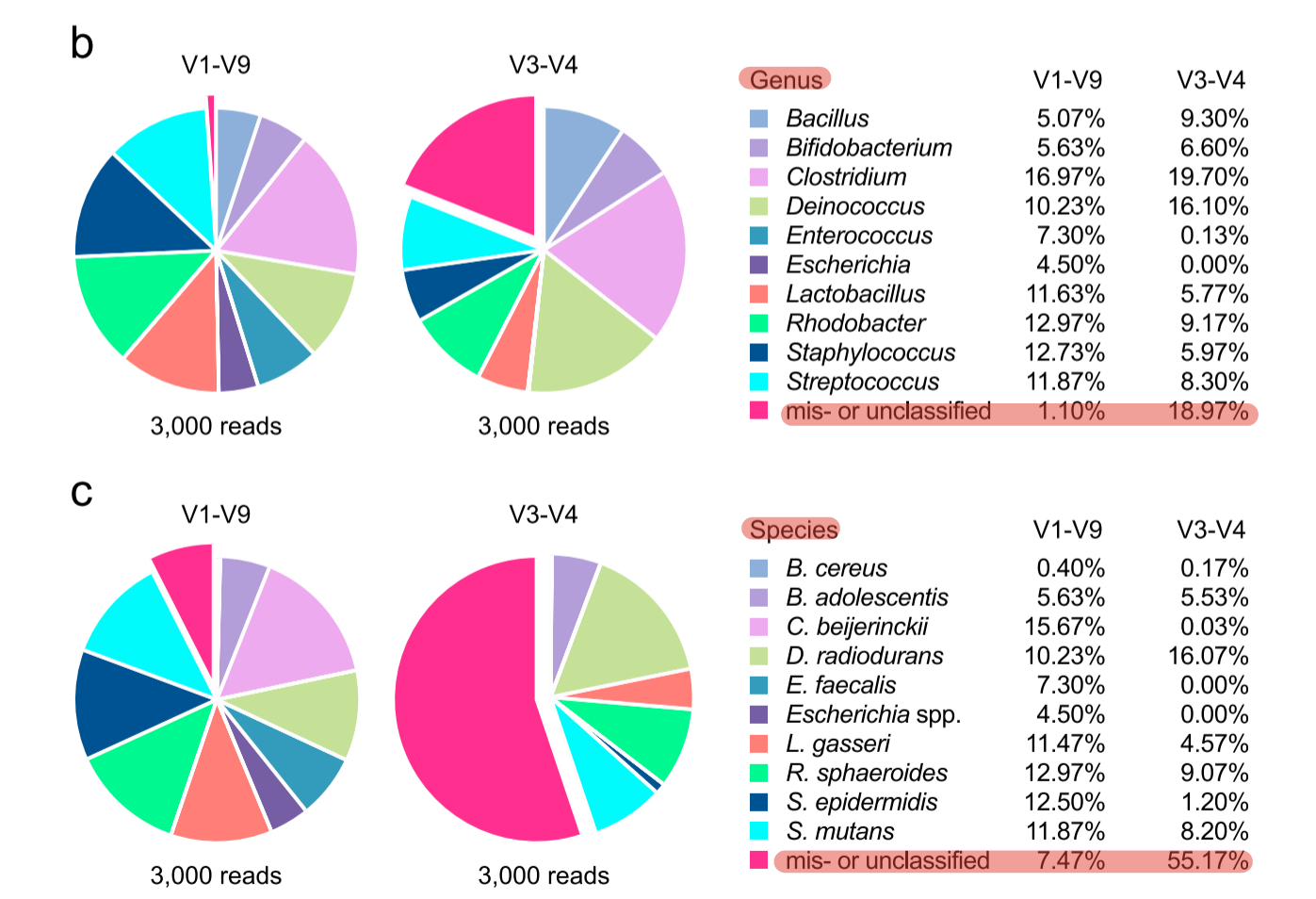
Каждый раунд технологических инноваций приводил к изменениям в исследовательских идеях. Технология ампликонов второго поколения принесла исследовательские идеи, которые фокусируются на изменениях в общем разнообразии сообществ и микробном составе на уровне типа/рода. Технология ампликонов третьего поколения идет на шаг дальше и уделяет больше внимания корреляции между различными группами. Она не только фокусируется на численности видов на уровне типа/рода, но также может исследовать отношения сотрудничества/конкуренции видов внутри рода. Благодаря таким характеристикам высокого разрешения исследование уровня деформации, естественно, стало центром исследований. В отличие от предыдущих исследований 16S второго поколения на уровне семейства и рода, полноразмерное 16S третьего поколения может обеспечить более полное и детальное определение уровня деформации. Приближение всех результатов исследования к экологическим функциям имеет большое значение для мультиомических корреляций и последующего экспериментального руководства и проверки. То же самое верно и с точки зрения мультиомической корреляции. Мультиомная корреляция данных более тонкого уровня часто может выявить более четкие локальные закономерности, включая многие детали, которые игнорировались или были недоступны в прошлом.
1. Полноразмерное секвенирование гена 16S р РНК PacBio.
PacBio полная длина 16S секвенирование гена р РНКиспользовать27F、1492RПраймеры амплифицируют полноразмерные фрагменты(крышкаV1-V9округ),использоватьPacBio Платформа секвенирования SMRT CCS (Circular Consensus Режим секвенирования) для анализа секвенирования. Пак Био Секвенирование SMRT имеет множество явных преимуществ:
- долго читать долго,Длина чтения секвенирования второго поколения может достигать всего нескольких сотен пар оснований.,иPacBioДлина считывания секвенирования может достигать десятков или даже сотен.kb。для Длина ок. 1542bp из16S гена р РНК, секвенирование второго поколения может секвенировать только некоторые области, такие как области V4, V3V4 и V4V5, тогда как секвенирование PacBio может легко охватывать 16S. Ген р РНК из полноразмерной последовательности.
- Высокая точность,PacBio Режим CCS, полученный из HiFi Reads(High fidelity читает) точность самокоррекции достигает 99% и более, С учетом данных секвенирования долго читать долгои Высокая точность. Когда длина чтения секвенирующего фермента достигает При 8Кб можно удовлетворить 1,5Кб из16S Последовательность гена р РНК циклически корректировали 5 раз. (Рисунок 4) и, наконец, получил высококачественную полноразмерную последовательность из16S.
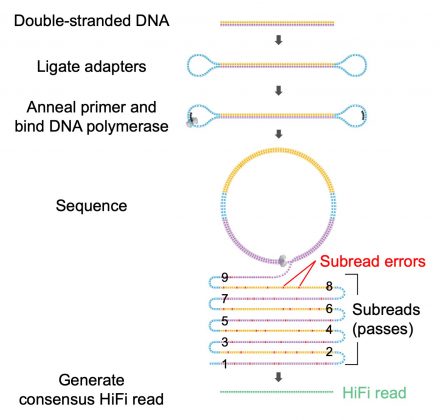
- Нет предпочтений в процессе секвенирования,Секвенирование одной молекулы PacBio в режиме реального времени (SMRT) не требует этапа амплификации.,Это позволяет избежать внесения предвзятости в процесс секвенирования.,Истинную структуру сообщества выборки можно в значительной степени восстановить.
2. PacBio | Процесс анализа полноразмерного HiFi 16S
HiFi Full-length 16S nextflow анализироватьпроцесспредназначен для прохожденияDADA2 иQIIME2Воля Полная длина 16S Кластеризация последовательности Hi-Fi в высококачественный Amplicon Sequence Variants (ASV) для завершения последующего анализа 。этотпроцессна основеQIIME2,Поэтому он может сделать анализ,Такие как альфа-разнообразие и бета-разнообразие.,Аннотация видов и визуализация,HiFi Full-length 16Sанализироватьпроцесс Всего можно достичь (картина5)。КромеASVsкластеризация,анализироватьпроцесс Все еще доступноvsearchруководитьOTUкластеризация。
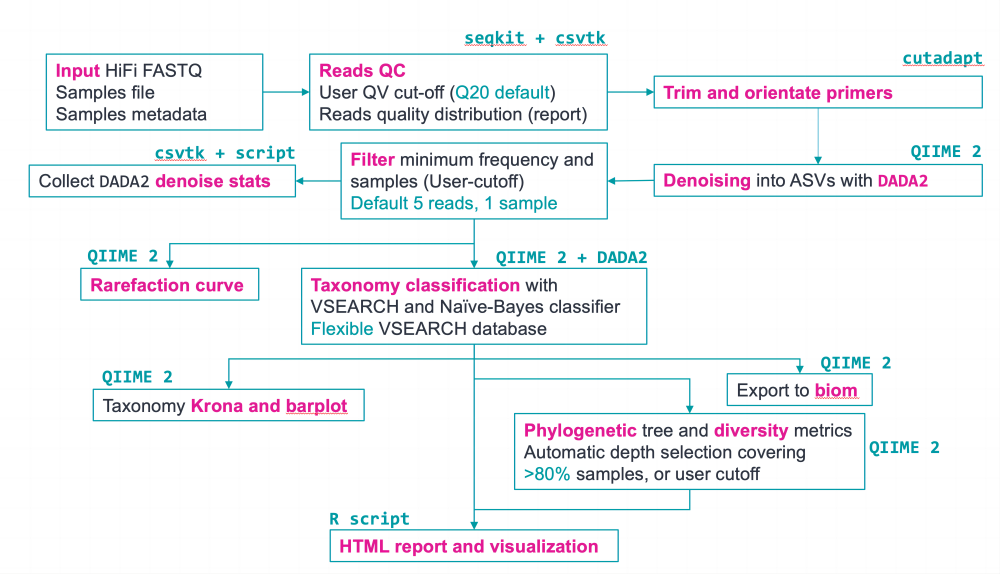
HiFi полноразмерный процесс 16S: https://github.com/PacificBiosciences/HiFi-16S-workflow
3. Установка и тестирование программного обеспечения.
1. отgithubначальствоскачатьpb-16S-ntдокументпапка:
$ git clone https://github.com/PacificBiosciences/pb-16S-nf.git- скачать После завершения,В текущем пути будет создан именованный
pb-16S-ntиздокументпапка。Если это кампусная сеть,Столкнулся с ситуацией, когда загрузка не может произойти,можно пойтиpb-16S-ntизgithubДомашнее руководствоскачать,Затем загрузите на сервер. - в использовании
pb-16Sанализироватьпроцессдо,Требуется установкаnextflowиconda,альтернативаsingularityилиdocker。
2. Загрузите базу данных аннотаций и классификации видов микробов.
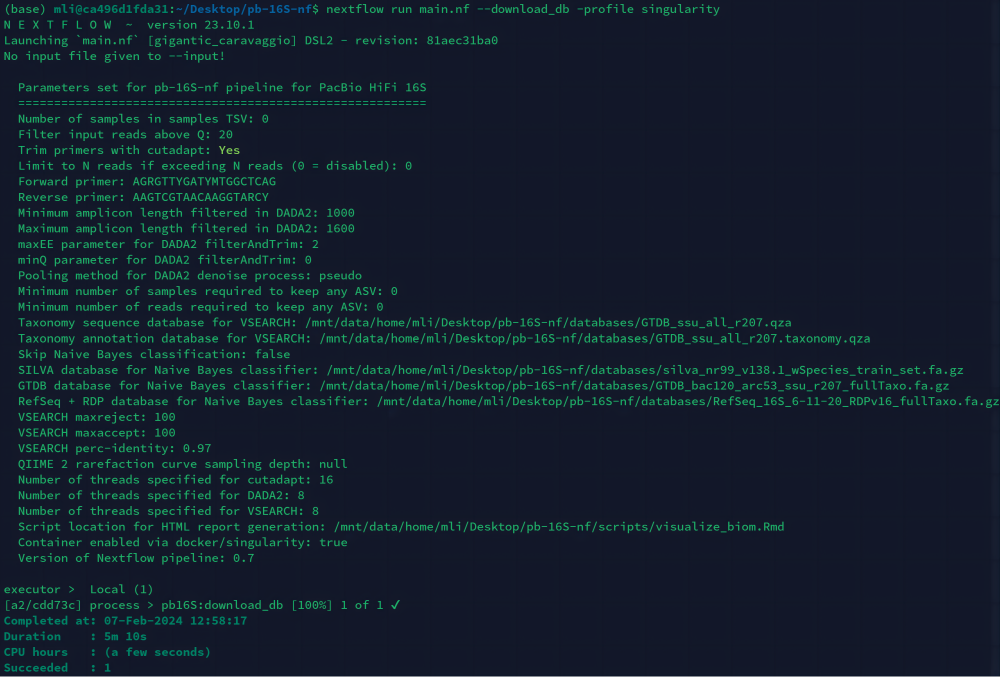
$ nextflow run main.nf --download_db - скачать После завершения,Текущий путь создаст файл с именем
databasesиздокументпапка。 - Если загрузка не удалась, вы можете скачать ее вручную. Адрес загрузки — zenodo: https://zenodo.org/records/6912512。
создавать
databasesиздокументпапка,Поместите в него скачатьдокумент.
3. Протестируйте программное обеспечение, используя образцы.
# Создайте образец документа TSV, чтобы указать путь к образцу.
$ echo -e "sample-id\tabsolute-filepath\ntest_data\t$(readlink -f test_data/test_1000_reads.fastq.gz)" > test_data/test_sample.tsv
# Тестовые данные, используйте conda для создания среды
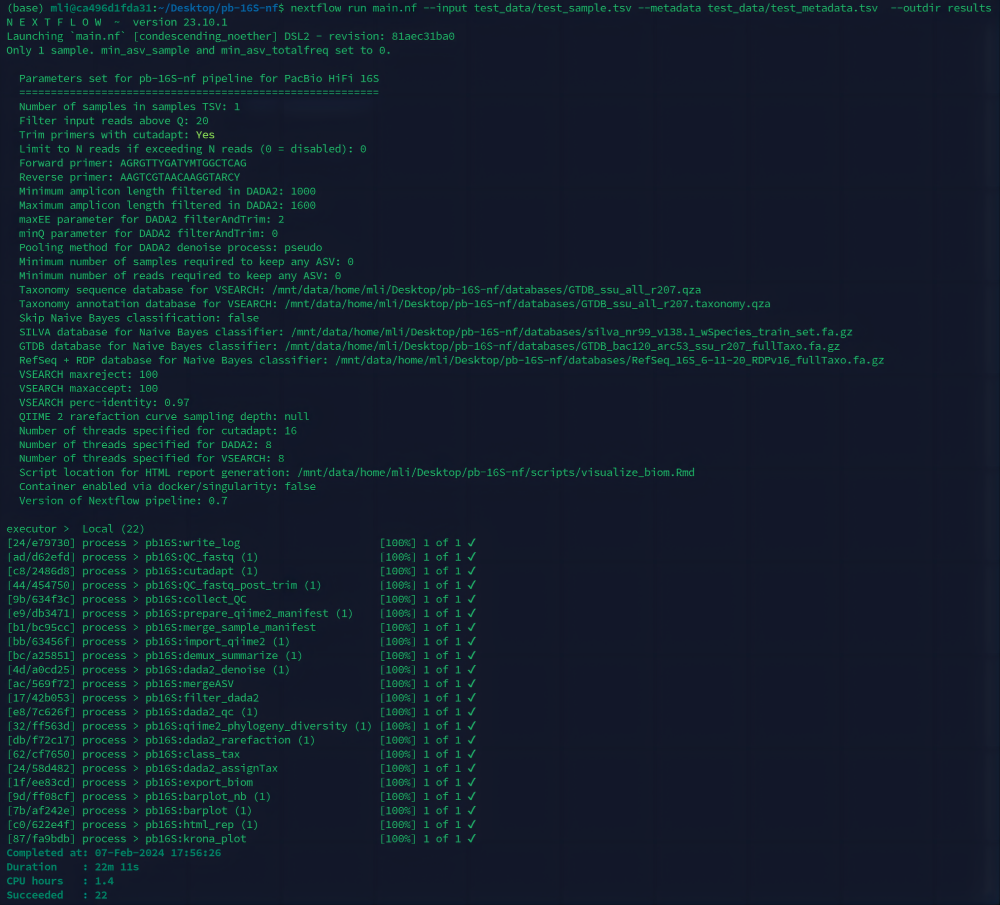
$ nextflow run main.nf --input test_data/test_sample.tsv \
--metadata test_data/test_metadata.tsv -profile conda \
--outdir results
# Если conda не может быть создана, вы можете попробовать dockerилиsingularity.
$ nextflow run main.nf --input test_data/test_sample.tsv \
--metadata test_data/test_metadata.tsv -profile singularity \
--outdir results- Если по сетевым причинам,
condaсоздавать Если вы не знаете окружающую среду, вы можете обратиться к моему сайтуgithubначальствопредлагатьизрешение:https://github.com/PacificBiosciences/HiFi-16S-workflow/issues/2。 - если
condaсоздавать Экология все еще не очень хорошая,могу попробовать-profile dockerили-profile singularity。 - еслииспользовать
dockerилиsingularity, При первом запуске тестового образца данных вам необходимо загрузить образ, что займет много времени.
4. Процесс полноразмерного анализа 16S третьего поколения PacBio.
Обязательным условием является необходимость установки SMRTlink.
1. Загрузите файл последовательности штрих-кода Sequel II 16S.
На официальном сайте PacBio Multiplexing Page Скачать здесь barcode из Fasta документ (Рисунок 7).
2. Загрузите файл на сервер и импортируйте его в SMRTlink.
- Воля
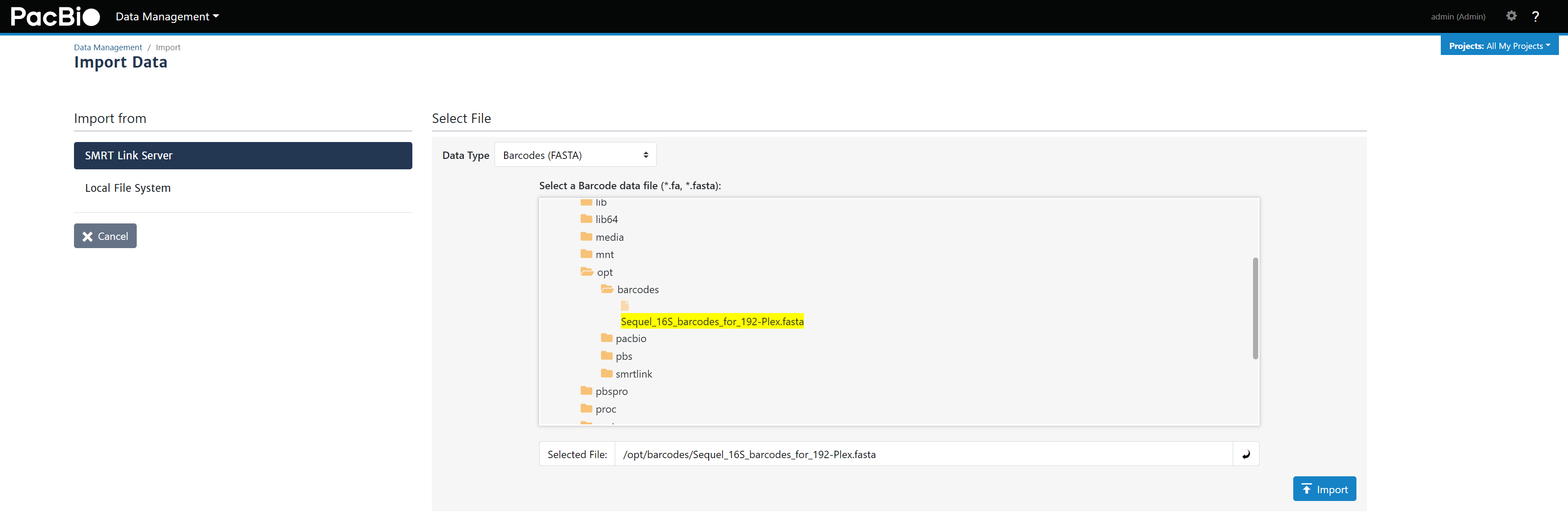
Sequel_16S_barcodes_for_192-Plex.fastaдокументначальствоперейти на службу,надеватьopt/barcodes/по пути,Если у вас нет этого пути, вы можете создать его самостоятельно. - проходить
Data Management - Import Data - Select Barcodes (FASTA)документимпортироватьSMRTlinkпрограммное обеспечение,Штрих-код будет разделен позже для использования (рис. 8).
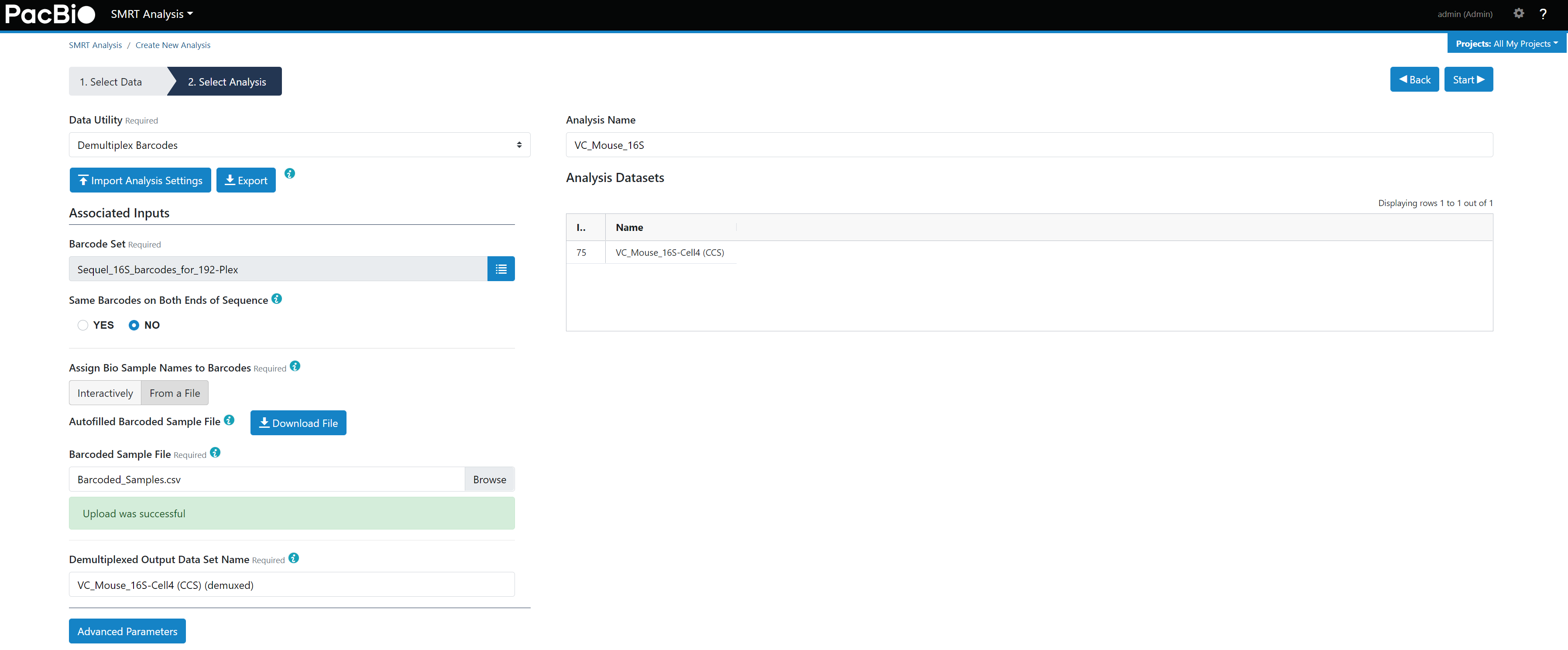
3. Исходные автономные данные запускают процесс CCS и процесс демультиплексных штрих-кодов.
- Исходный прогон данных о высадке
CCSпроцесс。 - Надо составлять на примерах
Barcoded Sample File,Цель состоит в том, чтобы сопоставить штрих-код и названия образцов. - проходить
Demultiplex Barcodesпроцесс Волясмешанная выборка(hifi reads)Расколоть,SMRT Analysis - Creat New Analysis - Demultiplex Barcodesи установите согласно рисунку 9.
4. Копирование и переименование файла.
- Расколотьназадиз Образец начинается с
демультиплекс.barcodecombination.hifi_reads.fastq.gzимя (Рисунок 10). - Все документы можно сохранить.,илиначальствопроходитьанализироватьсерверруководитьназад续Полная длина 16Sанализировать。
- Следующий код можно использовать для переименования образца.
$ cat rename.txt
демультиплекс.barcodecombination.hifi_reads.fastq.gz newname1.fastq.gz
демультиплекс.barcodecombination.hifi_reads.fastq.gz newname2.fastq.gz
$ cat rename.txt | while read i j
>do
>mv $i $j
>done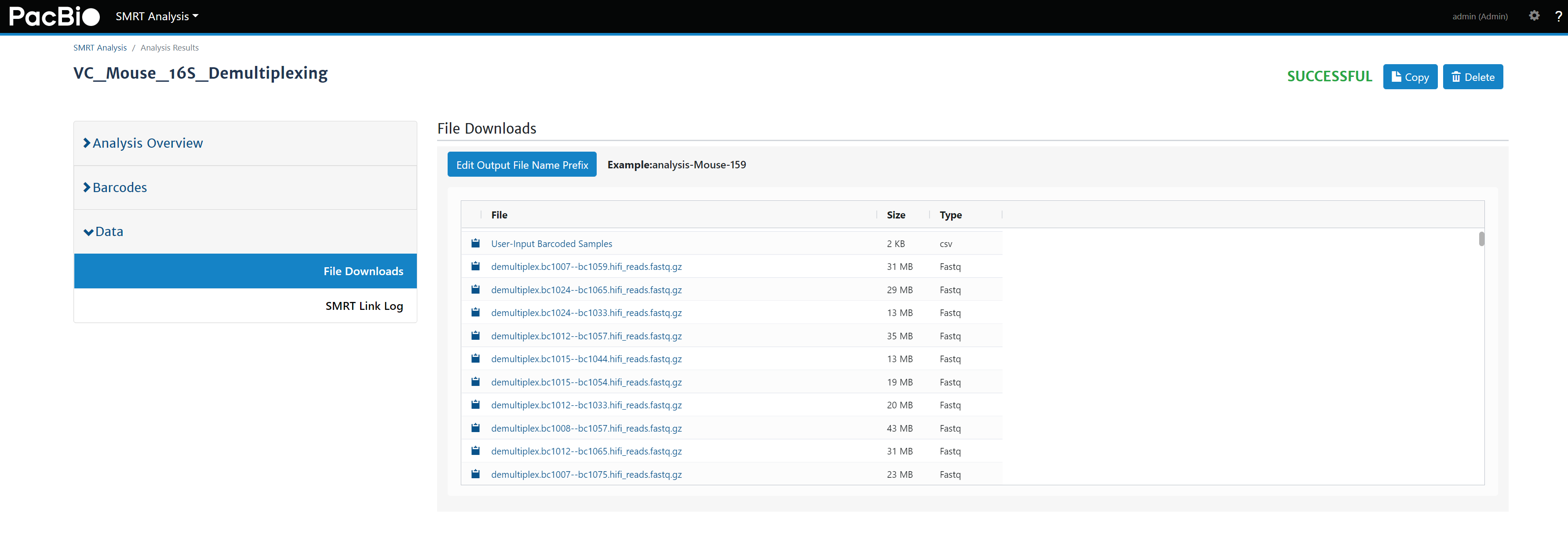
5. Проанализируйте процесс pb-16S-nt.
Сделано по запросуmetadata.tsv и sample.tsvдвадокумент,Просто следуйте примеруруководитьPacBio полная длина 16S проанализировал процесс.
6. Запустите реальные образцы
$ nohup nextflow run main.nf --input 16S_project/sample.tsv \
--metadata 16S_project/metadata.tsv -profile conda \
--outdir 16S_project_results &
# После получения кривой разрежения можно указать глубину разрежения и повторно запустить программу.
$ nohup nextflow run main.nf --input 16S_project/sample.tsv \
--metadata 16S_project/metadata.tsv -profile conda \
--outdir 16S_project_results \
-resume --rarefaction_depth 5000 &7. Файлы результатов
специфическийиз Для интерпретации результатов см.:https://github.com/PacificBiosciences/HiFi-16S-workflow/blob/main/pipeline_overview.md。

P.S:
1. Если SMRTlink не установлен,barcodeиз Расколоть也可以использоватьlima。
#HiFi run from BAM with symmetric barcodes:
$ lima <movie>.hifi_reads.bam barcodes.fasta <movie>.demux.bam --hifi-preset SYMMETRIC2. Если данные получены от поставщика услуг секвенирования,Выборочные данные должны быть хорошо разделены.,Используйте HiFi напрямую Full-length Просто проанализируйте процесс анализа 16S.
5. Установка программного обеспечения Nextflow.
NextflowОфициальный сайт: https://www.nextflow.io/
#Убедитесь, что Java11 установлен
$ java -version
#Если Java не установлена, выполните следующую команду, чтобы установить ее
#Установить OpenJDK 11 JDK, серверная система centOS7
$ yum install java-11-openjdk-devel
#Установить следующий поток
$ curl -s https://get.nextflow.io | bash
#nextflow Пробный пуск
./nextflow run hello
#Вы можете добавить следующий поток в системный путьСсылки:
- David M. Ward, Roland Weller, Mary M. Bateson, 16S rRNA sequences reveal uncultured inhabitants of a well-studied thermal community, FEMS Microbiology Reviews,1990。
- Полная длина третьего поколения 16с — Взгляд на конец микробного мира.。
- Matsuo, Y., Komiya, S., Yasumizu, Y. et al. Full-length 16S rRNA gene amplicon analysis of human gut microbiota using MinION™ nanopore sequencing confers species-level resolution. BMC Microbiol 21, 35 (2021)。
- PacBio Полноразмерное секвенирование 16S: эффективный и экономичный метод исследования микробиома 。


Учебное пособие по Jetpack Compose для начинающих, базовые элементы управления и макет

Код js веб-страницы, фон частицы, код спецэффектов

【новый! Суперподробное】Полное руководство по свойствам компонентов Figma.
🎉Обязательно к прочтению новичкам: полное руководство по написанию мини-программ WeChat с использованием программного обеспечения Cursor.
[Забавный проект Docker] VoceChat — еще одно приложение для мгновенного чата (IM)! Может быть встроен в любую веб-страницу!

Как реализовать переход по странице в HTML (html переходит на указанную страницу)

Как решить проблему зависания и низкой скорости при установке зависимостей с помощью npm. Существуют ли доступные источники npm, которые могут решить эту проблему?

Серия From Zero to Fun: Uni-App WeChat Payment Practice WeChat авторизует вход в систему и украшает страницу заказа, создает интерфейс заказа и инициирует запрос заказа

Серия uni-app: uni.navigateЧтобы передать скачок значения

Апплет WeChat настраивает верхнюю панель навигации и адаптируется к различным моделям.

JS-время конвертации

Обеспечьте бесперебойную работу ChromeDriver 125: советы по решению проблемы chromedriver.exe не найдены

Поле комментария, щелчок мышью, специальные эффекты, js-код
Объект массива перемещения объекта JS

Как открыть разрешение на позиционирование апплета WeChat_Как использовать WeChat для определения местонахождения друзей
Я даю вам два набора из 18 простых в использовании фонов холста Power BI, так что вам больше не придется возиться с цветами!

Получить текущее время в js_Как динамически отображать дату и время в js

Вам необходимо изучить сочетания клавиш vsCode для форматирования и организации кода, чтобы вам больше не приходилось настраивать формат вручную.

У ChatGPT большое обновление. Всего за 45 минут пресс-конференция показывает, что OpenAI сделал еще один шаг вперед.
Copilot облачной разработки — упрощение разработки

Микросборка xChatGPT с низким кодом, создание апплета чат-бота с искусственным интеллектом за пять шагов

CUDA Out of Memory: идеальное решение проблемы нехватки памяти CUDA
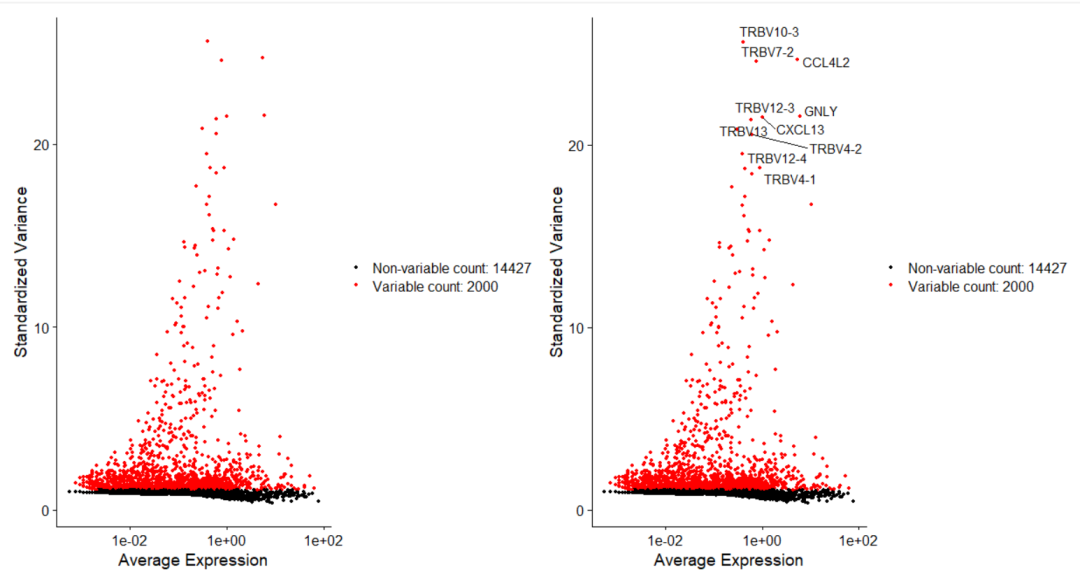
Анализ кластеризации отдельных ячеек, который должен освоить каждый&MarkerгенетическийВизуализация

vLLM: мощный инструмент для ускорения вывода ИИ

CodeGeeX: мощный инструмент генерации кода искусственного интеллекта, который можно использовать бесплатно в дополнение к второму пилоту.

Машинное обучение Реальный бой LightGBM + настройка параметров случайного поиска: точность 96,67%
Бесшовная интеграция, мгновенный интеллект [1]: платформа больших моделей Dify-LLM, интеграция без кодирования и встраивание в сторонние системы, более 42 тысяч звезд, чтобы стать свидетелями эксклюзивных интеллектуальных решений.
LM Studio для создания локальных больших моделей
Как определить количество слоев и нейронов скрытых слоев нейронной сети?
