Обучение и сортировка процесса Scanpy с одной ячейкой (10-кратное считывание/фильтрация/уменьшение размерности/кластеризация одной выборки)
Я планирую внимательно изучить структуру корреляционного анализа отдельных ячеек, основанную на Python, ха-ха.
Написание кода очень утомительно для новичков, поэтому, пожалуйста, будьте терпеливы. В этот раз я использовал среду разработки Visual Studio.
Этот процесс взят с официального сайта Scanpy (предварительная обработка и кластеризация 3000 PBMC (устаревший рабочий процесс)):
https://scanpy.readthedocs.io/en/stable/tutorials/basics/clustering-2017.html
Шаг процесса
1、check
# текущий рабочий каталог
import os
# текущий рабочий каталог
os.getcwd()
# '/Users/zaneflying/Desktop/scanpy'В Python os — это стандартная библиотека, предоставляющая ряд функций для взаимодействия с операционной системой. Этот пакет позволяет выполнять такие задачи, как операции с файлами и каталогами (например, создание, удаление, изменение), получать информацию, связанную с операционной системой, обрабатывать пути к файлам и т. д. Это одна из основных библиотек Python, которая не требует дополнительной установки, ее можно импортировать и использовать напрямую. Это делает пакет os очень полезным инструментом при решении задач файловой системы и операционной системы.
# Файлы в каталоге
os.listdir()
# ['scanpy.ipynb', '.DS_Store', 'input', 'data']2. Импорт
Кстати, форма установки библиотеки такая: pip install pandas
import pandas as pd
import scanpy as sc
import numpy as np
import anndata as ad
import poochPandas — это библиотека анализа данных Python с открытым исходным кодом, которая широко используется для быстрого анализа, очистки и подготовки данных. Он предоставляет эффективные объекты DataFrame, упрощая создание, манипулирование и предварительную обработку структурированных данных в Python. Pandas предоставляет широкий спектр простых в использовании функций и методов для импорта, преобразования, очистки и визуализации данных.
3. Установите параметры перед анализом.
sc.settings.verbosity = 3
sc.logging.print_header()
# scanpy==1.10.3 anndata==0.10.9 umap==0.5.6 numpy==2.0.2 scipy==1.13.1 pandas==2.2.3 scikit-learn==1.5.2 statsmodels==0.14.3 igraph==0.11.6 louvain==0.8.2 pynndescent==0.5.13
sc.settings.set_figure_params(dpi=80, facecolor="white")Изучите приведенный выше код:
sc.settings.verbosity?
# Добавьте ? после функции, а затем запустите ее, чтобы просмотреть справочный документ по функции.
# Давайте разберем функцию этой функции. Это сокращение от scdascanpy, за которым следует settings.verbosity. Что касается детального уровня ведения журнала, 3 означает отображение подсказок.
sc.logging.print_header?
# Эта функция используется для печати информации о версии различных зависимых пакетов, которая может повлиять на числовые результаты.
sc.settings.set_figure_params?
# sc.settings.set_figure_params(
# *,
# scanpy: 'bool' = True,
# dpi: 'int' = 80,
# dpi_save: 'int' = 150,
# frameon: 'bool' = True,
# vector_friendly: 'bool' = True,
# fontsize: 'int' = 14,
# figsize: 'int | None' = None,
# color_map: 'str | None' = None,
# format: '_Format' = 'pdf',
# facecolor: 'str | None' = None,
# transparent: 'bool' = False,
# ipython_format: 'str' = 'png2x',
# ) -> 'None'
# Эта функция устанавливает размер изображения, который сильно отличается от языка R. Обычно язык R устанавливает его на этапе вывода.
# scanpy: Если это правда, используйте настройки matplotlib Scanpy по умолчанию.
# dpi: Разрешение, с которым отображается изображение.
# dpi_save: Разрешение, в котором сохраняется изображение.
# frameon: да Отображать ли рамку вокруг изображения.
# vector_friendly: да Следует ли генерировать векторные выходные данные.
# fontsize: Размер шрифта, используемый в графике.
# figsize: Размер изображения. Если нет, используется размер по умолчанию.
# color_map: Цветовая карта, которую нужно использовать.
# format: Формат, используемый при сохранении рисунка.
# facecolor: Цвет фона изображения. Если «Нет», используется цвет по умолчанию.
# transparent: да Делать ли графический фон прозрачным.
# ipython_format: В Юпитере Формат, используемый при отображении графики в Notebook.3. Установите путь вывода файла.
# Установить путь к файлу
results_file = "output/" # the file that will store the analysis results4. Чтение файлов
adata = sc.read_10x_mtx(
"input/", # the directory with the `.mtx` file
var_names="gene_symbols", # use gene symbols for the variable names (variables-axis index)
cache=True, # write a cache file for faster subsequent reading
)Перейдите в папку, где хранятся данные, и выберите подходящий метод чтения. Например, автор использует данные 10X, поэтому я выбираю для чтения sc.read_10x_mtx.
Помимо этого, есть много других способов чтения.
## Preprocessing
sc.read_10x_mtx?
# path (Path | str):
# Указывает путь к каталогу файлов, включая .mtx и .tsv документ. Например './filtered_gene_bc_matrices/hg19/'。
# var_names (Literal['gene_symbols', 'gene_ids'], optional, default: 'gene_symbols'):
# Способ определения индекса переменной вы можете выбрать 'gene_symbols' или 'gen_ids'. По умолчанию да 'gene_symbols'。
# make_unique (bool, optional, default: True):
# да Нет гарантирует, что индекс переменной уникален (путем добавления дубликата '-1', '-2' ждать).Значение по умолчаниюда True。
# cache (bool, optional, default: False):
# да Кэшировать ли прочитанные файлы. Значение по умолчаниюда False。
# cache_compression (Literal['gzip', 'lzf'] | None | Empty, optional):
# Указывает формат сжатия кэша (например. 'gzip' или 'lzf'), значение по умолчанию: _empty, то есть не указано.
# gex_only (bool, optional, default: True):
# если для Правда, только экспрессия генов (ген expression, GEX) данные. Значение по умолчаниюда True。
# prefix (str | None, optional):
# Вы можете добавить префикс перед именем файла, по умолчанию: None。
# возвращаемое значение функции
# возвращаемое значение (AnnData):
# Функция возвращает AnnData Объект, общая структура, используемая для хранения данных многомерного массива, обычно для отдельных ячеек. RNA-seq анализ данных。контрольные данные
adataAnnData object with n_obs × n_vars = 8931 × 33538:
n_obs: представляет количество наблюдений, то есть количество клеток в данных одноклеточной РНК-секвенирования. В этом примере имеется 8931 ячейка (n_obs = 8931).
n_vars: представляет количество переменных, то есть количество измеряемых генов. В этом примере имеется 33538 генов (n_vars = 33538).
В совокупности это представляет собой матрицу экспрессии, состоящую из 8931 клеток × 33538 генов.
var: 'gene_ids', 'feature_types':
var — это информация аннотации, относящаяся к переменной (гену) в объекте AnnData. Здесь перечислены два поля аннотации:
'gene_ids': идентификатор гена, соответствующий каждому гену.
'feature_types': особенности типов генов (например, гены, кодирующие белки, некодирующие РНК и т. д.).
Эти поля аннотаций хранят метаданные о каждом гене и могут использоваться для фильтрации, группировки и других операций в процессе анализа.
5. Сделайте это уникальным именем гена
adata.var_names_make_unique()Если это Gene_ids, вам не нужно делать этот шаг, но его рекомендуется добавить всем. Автор здесь использует Gene_symbols
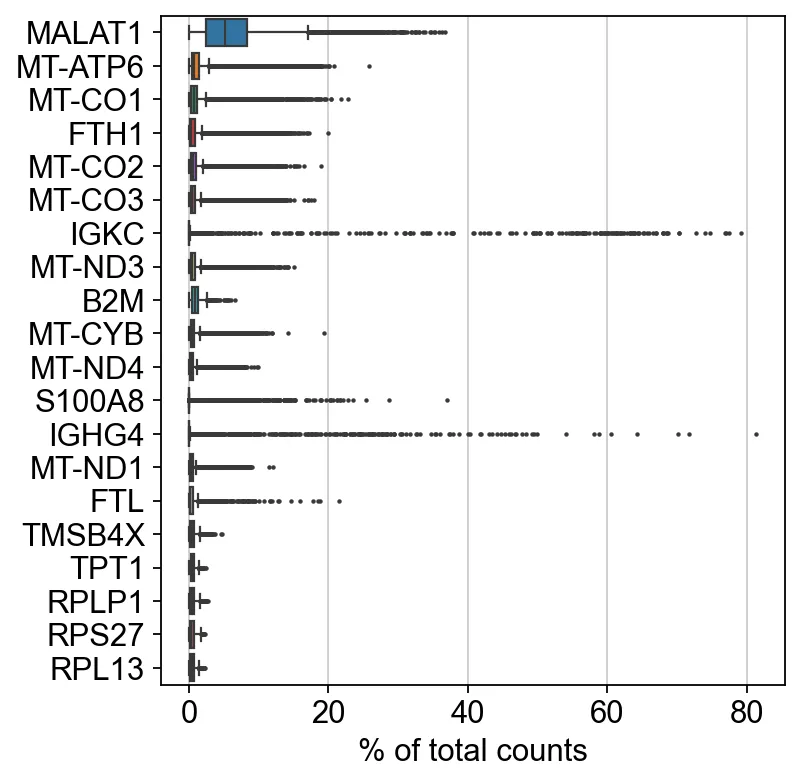
6. Найдите 20 самых высокоэкспрессируемых генов.
sc.pl.highest_expr_genes(adata, n_top=20)sc означает scanpy, а pl означает сюжет. Этот код можно использовать во многих ситуациях
7. Фильтровать данные
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
# filtered out 196 cells that have less than 200 genes expressed
# filtered out 11773 genes that are detected in less than 3 cells8. Добавьте список митохондриальных генов.
adata.var["mt"] = adata.var_names.str.startswith("MT-")
sc.pp.calculate_qc_metrics(
adata, qc_vars=["mt"], percent_top=None, log1p=False, inplace=True
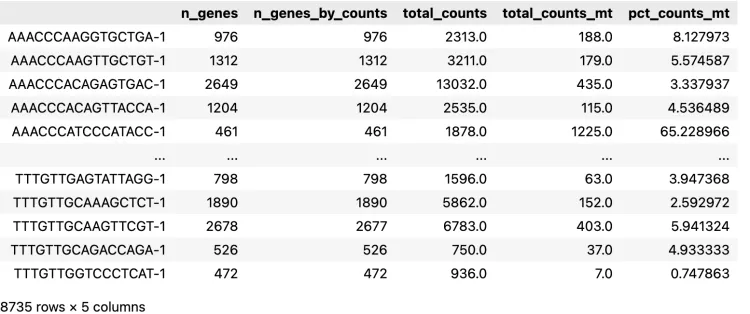
)adata.obsПроверьте данные наблюдения
adata.var_names.str.startswith?
# adata даон AnnData вернослон,Он используется для хранения и управления отдельными ячейками. RNA Основная структура данных секвенирования.
# var_names да adata Объектатрибут,Указывает имя гена в наборе данных или ID(то есть имя переменной)。Конкретно,этодаон pandas.Index вернослон,Содержит названия всех генов или ID。
# .str да pandas Предоставляет метод доступа для методов манипулирования строками, используемый для pandas.Series или Index Выполните операции векторизации над строками в файлах .
# проходить .str, ты можешь var_names Все имена генов (строки) в одновременно подвергаются различным строковым операциям, таким как проверка префиксов, суффиксов, отношений включения, замена подстрок и т. д.
# .startswith даон Строковые методы,для проверки строкида Нет начинается с указанного префикса。
# Когда с .str При совместном использовании начинается с можно использовать для проверки var_names Имя каждого гена да в генах начинается с определенного префикса.sc.pp.calculate_qc_metrics?
# qc_vars Параметр указывает категорию генов, которая будет использоваться для контроля качества. Обычно это список.,Содержит префикс, используемый для идентификации определенного набора генов.илизакрывать键字。
# Здесь, ["мт"] Указывает на митохондриальные гены (обычно "MT-" или Гены, начинающиеся с похожих префиксов)。этот意味着该функция将计算与线粒体基因相закрывать的 QC индекс.
# Этот параметр указывает префикс, который будет рассчитан. n На долю наиболее экспрессируемых генов приходится процент от общей экспрессии. Например, процент_топ=[50, 100] Сначала посчитаем наиболее выраженный 50 До 100 Общий процент экспрессии генов.
# Установить здесь, чтобы None,означает не считать этот индекс.
# log1p Параметр определяет, рассчитывается ли да. QC Логарифмическое преобразование данных перед индикатором (log(1 + x))。
# Здесь log1p=False Указывает, что данные не будут логарифмически преобразованы.
# Этот параметр определяет, является ли функция да прямым входом. adata Добавьте рассчитанное QC индекс.
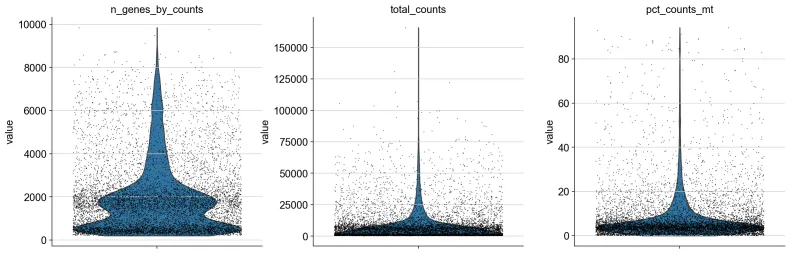
# inplace=True представляет собой рассчитанный QC Индикаторы будут добавлены непосредственно в adata вернослон中,Вместодавернуть новыйвернослон。9. Чертеж схемы скрипки.
sc.pl.violin(
adata,
["n_genes_by_counts", "total_counts", "pct_counts_mt"],
jitter=0.4,
multi_panel=True,
)
sc.pl.violin?
# ["n_genes_by_counts", "total_counts", "pct_counts_mt"]:
# этотдаон字符串список,指定要绘制的小提琴картина的Изменять量名称:
# n_genes_by_counts: 每клетки Количество обнаруженных генов(Обычно относится к числу генов с ненулевой экспрессией.)。этотда Мера клеточной сложностииндекс.
# total_counts: Всего на клетку UMI(Unique Molecular Идентификатор) подсчет, обозначающий общее количество RNA количество.
# pct_counts_mt: Экспрессия митохондриальных генов в процентах от общей экспрессии,Обычно используется для обнаружения мертвых клеток и клеток низкого качества.
# Эти переменные обычно генерируются на этапах контроля качества (КК).
# jitter=0.4:
# jitter Параметры управляют расположением точек на диаграмме рассеяния. x Джиттер на валуколичество.Дизеринг предотвращает перекрытие точек.,Сделайте распределение данных более видимым。
# Здесь джиттер = 0,4 Указывает на небольшое разнесение точек во избежание перекрытия.
# multi_panel=True:
# multi_panel Параметр определяет, отображает ли несколько переменных на разных панелях графиков скрипки.
# multi_panel=True Указывает, что каждая переменная отображается на отдельной панели графика скрипки вместо отображения всех переменных на одном графике.10. Рисунок диаграммы рассеяния
sc.pl.scatter(adata, x="total_counts", y="pct_counts_mt")
sc.pl.scatter(adata, x="total_counts", y="n_genes_by_counts")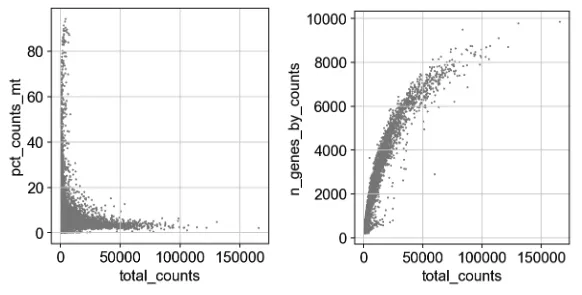
11. Дальнейшая фильтрация
Отфильтруйте всю информацию с количеством 2500 и процентом митохондрий более 5.
Из пояснения, использование .copy не повлияет на данные перед этим шагом (у автора здесь нет четкого понимания, но смысл именно в этом)
adata = adata[adata.obs.n_genes_by_counts < 2500, :]
adata = adata[adata.obs.pct_counts_mt < 5, :].copy()
adata = adata[adata.obs.n_genes_by_counts < 2500, :]:
# adata.obs.n_genes_by_counts:этотдаон Содержит всеклетки中Количество обнаруженных генов的列,хранится в AnnData Объект obs в кадре данных.
# adata[adata.obs.n_genes_by_counts < 2500, :]: Функция этой строки кода — отфильтровать те гены, количество обнаруженных генов которых меньше 2500 клетки.
# adata.obs.n_genes_by_counts < 2500 Создайте логический вектор, определяющий, какие клетки имеют меньше генов, чем 2500。
# adata[...] Используйте этот логический вектор, чтобы выбрать ячейки, соответствующие критериям, и удалить ячейки, которые не соответствуют.
# Результат: Число сохранившихся генов меньше 2500 ячейки, отфильтровывая другие ячейки.
# adata = adata[adata.obs.pct_counts_mt < 5, :].copy():
# adata.obs.pct_counts_mt:этотдаон Содержит всеклетки Столбец, показывающий долю экспрессии митохондриальных генов от общей экспрессии в。
# adata[adata.obs.pct_counts_mt < 5, :]: Эта строка кода дополнительно отфильтровывает митохондриальные гены, коэффициент экспрессии которых меньше 5% клетки.
# adata.obs.pct_counts_mt < 5 Генерирует логический вектор, определяющий, в каких клетках доля экспрессии митохондриальных генов меньше, чем 5%。
# adata[...] Используйте этот логический вектор, чтобы выбрать ячейки, соответствующие критериям, и удалить ячейки, которые не соответствуют.
# .copy(): Этот метод создает adata Объекткопировать,Убедитесь, что последующие операции не влияют на исходные данные.12. Нормализовать данные
Параметры этого кода очень похожи на параметры в seruat.
sc.pp.normalize_total(adata, target_sum=1e4)13. После нормализации вам все равно необходимо записать данные.
log1p — это сокращение от log(1 + x), что означает добавление 1 к каждому значению x и последующее взятие натурального логарифма.
sc.pp.log1p(adata)Seruat объединяет два вышеуказанных шага.
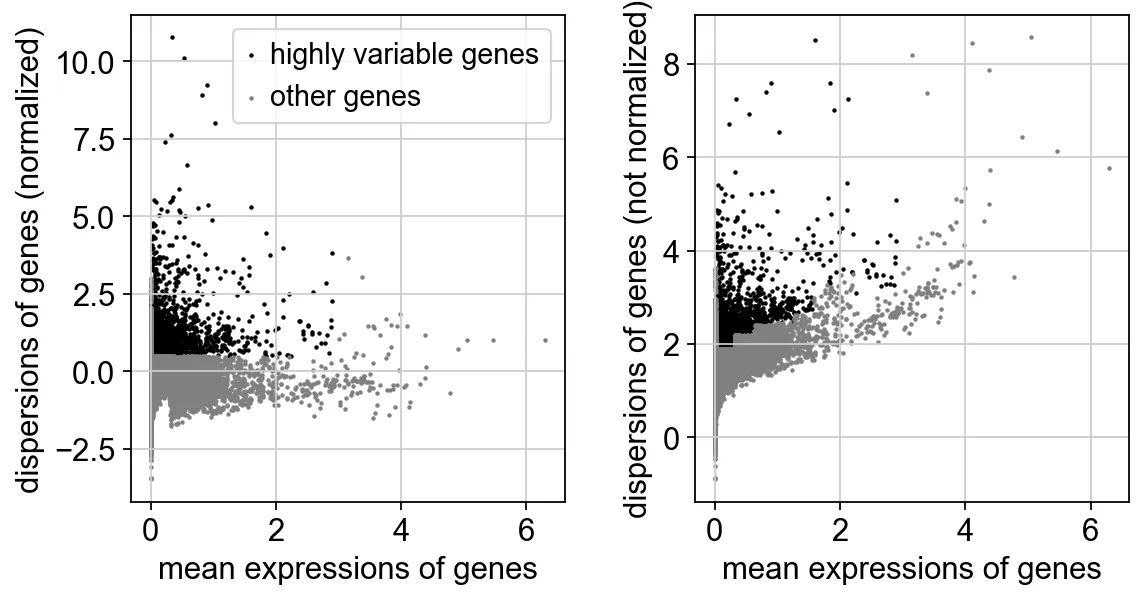
14. Найдите гипермутабельные гены.
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
# min_mean Параметр устанавливает минимальный средний порог экспрессии, при котором ген считается сильно вариабельным. Другими словами, только те, чье среднее значение экспрессии превышает 0.0125 гены будут рассматриваться как высоковариабельные гены-кандидаты.
# max_mean Параметр устанавливает максимальный средний порог экспрессии, при котором ген считается сильно вариабельным. Среднее значение экспрессии выше, чем 3 гены будут исключены.
# Обычно это делается, чтобы избежать выбора сверхэкспрессируемых генов, которые могут доминировать в анализе, но не представляют биологически значимых изменений.
# min_disp Параметр задает минимальный порог изменчивости (дисперсии) генов. Только те, изменчивость которых превышает 0.5 гены будут рассматриваться как высоковариабельные гены-кандидаты.
Изменять异度даотносится к фазе значения экспрессии генаверно Стандартизованная дисперсия среднего значения,Гены с более высокой вариабельностью экспрессируются по-разному в разных популяциях клеток.sc.pl.highly_variable_genes(adata)
# sc да scanpy Аббревиатура обозначает всю библиотеку.
# pl да plot аббревиатура,表示этотдаон Модули для рисованияилифункция。
# scanpy Библиотека построена иерархически по функциональным модулям, включая:
# sc.pp (pp да preprocessing (сокращение от ): используется для этапов предварительной обработки данных, таких как нормализация, фильтрация данных, идентификация сильно вариабельных генов и т. д.
# sc.tl (tl да tools аббревиатура): используется для вызова различных инструментов анализа, таких как PCA, кластеризация, UMAP, анализ дифференциальных выражений и т. д.
# sc.pl (pl да plot аббревиатура): используется для визуализации данных, например рисования. PCA Рисунок, УМАП картина、тепловая карта、Схема скрипки и т. д.Постройте диаграмму рассеяния сильно изменчивых генов
15. Масштабируйте данные
sc.pp.scale(adata, max_value=10)
# sc.pp.scale(adata, max_value=10) верно adata вернослон中的基因表达数据进行标准化,и ограничить стандартизированные данные [-10, 10] в пределах диапазона16. PCA-анализ, уменьшение размерности
sc.tl.pca(adata, svd_solver="arpack")Нарисуйте график PCA
sc.pl.pca(adata, color="CST3")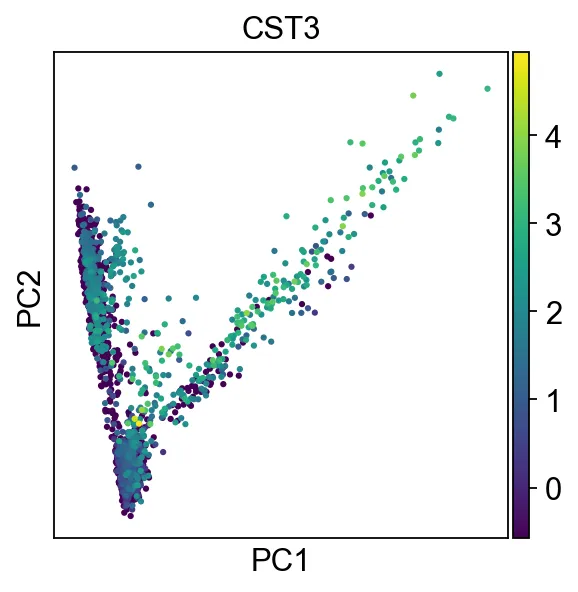
17. Отобразите вклад каждого главного компонента в данные.
sc.pl.pca_variance_ratio(adata, log=True)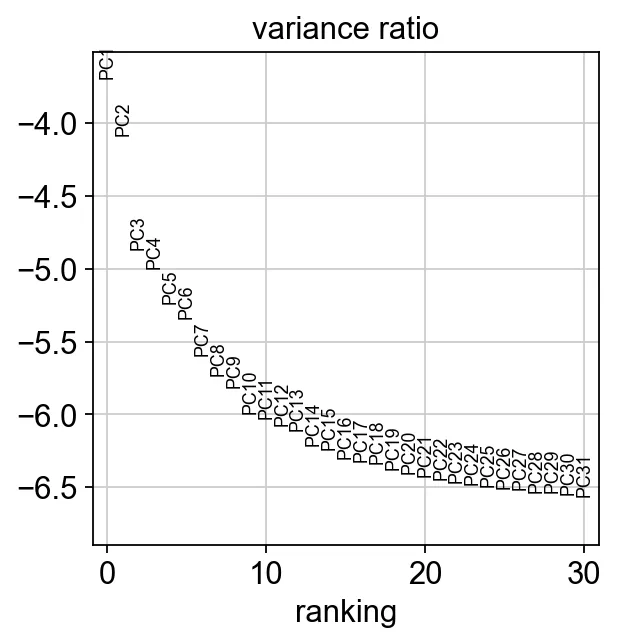
18. Рассчитать граф окрестности
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40)umap
sc.tl.umap(adata, init_pos='paga')sc.pl.umap(adata, color=["CST3", "NKG7", "PPBP"])Нарисуйте картинки umap на основе этих генов.
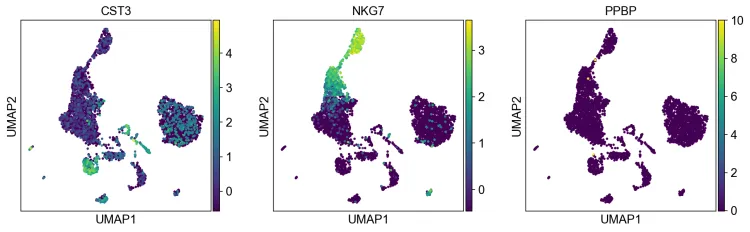
19. Кластеризация
sc.tl.leiden(
adata,
resolution=0.9,
random_state=0,
flavor="igraph",
n_iterations=2,
directed=False,
)
# random_state Параметры используются для установки случайных начальных чисел, чтобы обеспечить повторяемость результатов кластеризации. установлен на 0 Представляет с теми же данными и настройками,Запуск алгоритма несколько раз приведет к одному и тому же результату. Это необходимо для обеспечения согласованности результатов при многократном запуске.,Особенно, когда необходимо повторить эксперимент и поделиться результатами.
# flavor Параметр указывает, какая реализация используется. "график" даа тип на основе igraph Реализация библиотеки, это да Leiden Реализация алгоритма по умолчанию. iграф даон高效的картина处理库,通常用于картинаисетевой анализ。это支持快速的社区检测(нравиться Leiden кластеризация).
# n_iterations Параметр задает максимальное количество итераций алгоритма. Каждая итерация пытается оптимизировать модульность графа, тем самым улучшая разделение сообщества. n_итераций=2 Указывает на то, что наиболее 2 итерации,Часто используется для балансировки вычислительной эффективности.икластеризациякачество результатовколичество.нравиться果算法在早期迭代中已经收敛,Возможно, нет необходимости достигать максимального количества итераций.。
# directed Параметр указывает, является ли граф да ориентированным графом. Если установлено значение Неверно, он считается неориентированным графом. В большинстве одиночных клеток RNA-seq анализ В данных граф сходства ячеек обычно рассматривается как неориентированный граф, поэтому для этого параметра обычно устанавливается значение Falsesc.pl.umap(adata, color=["leiden"])Диаграмму кластеризации при разрешении=0,9 можно разделить на 17 кластеров.
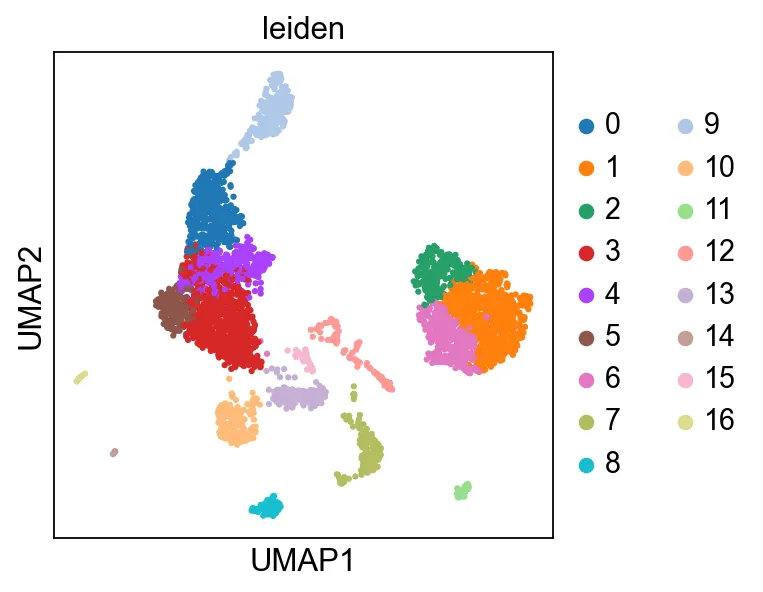
Очистите код
# Загрузить библиотеку
import pandas as pd
import scanpy as sc
import numpy as np
import anndata as ad
import pooch
# Настройки параметров
sc.settings.verbosity = 3
sc.logging.print_header()
sc.settings.set_figure_params(dpi=80, facecolor="white")
# Установить путь к файлу
results_file = "output/"
# Чтение данных
adata = sc.read_10x_mtx(
"input/",
var_names="gene_symbols",
cache=True,
)
# Не допускайте повторения названий генов
adata.var_names_make_unique()
# Базовая фильтрация
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
# Добавьте данные митохондриального гена
adata.var["mt"] = adata.var_names.str.startswith("MT-")
sc.pp.calculate_qc_metrics(
adata, qc_vars=["mt"], percent_top=None, log1p=False, inplace=True
)
# Чертеж основных данных
sc.pl.violin(
adata,
["n_genes_by_counts", "total_counts", "pct_counts_mt"],
jitter=0.4,
multi_panel=True,
)
# Чертеж основных данных
sc.pl.scatter(adata, x="total_counts", y="pct_counts_mt")
sc.pl.scatter(adata, x="total_counts", y="n_genes_by_counts")
# фильтр
adata = adata[adata.obs.n_genes_by_counts < 2500, :]
adata = adata[adata.obs.pct_counts_mt < 5, :].copy()
# Параметры фильтра можно настроить на основе приведенной выше информации.
##################################
# Нормализовать+записать данные
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
# В поисках гипервариабельных генов
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
# Гипервариабельное картирование генов
sc.pl.highly_variable_genes(adata)
# Сделать резервную копию
adata.raw = adata
# масштабировать данные
sc.pp.scale(adata, max_value=10)
# уменьшение размерности PCA
sc.tl.pca(adata, svd_solver="arpack")
# Рассчитать граф окрестности + обработка umap
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40)
sc.tl.umap(adata)
#################
# Лейденская кластеризация
sc.tl.leiden(
adata,
resolution=0.9,
random_state=0,
flavor="igraph",
n_iterations=2,
directed=False,
)Примечание:нравитьсяверно Есть сомнения в содержанииили У автора есть друзья, которые обнаружили явные ошибки.,Пожалуйста, свяжитесь с серверной частью(Добро пожаловать для общения)。更多内容可закрывать Примечание Официальный аккаунт:Ковчег рождения
- END -


Учебное пособие по Jetpack Compose для начинающих, базовые элементы управления и макет

Код js веб-страницы, фон частицы, код спецэффектов

【новый! Суперподробное】Полное руководство по свойствам компонентов Figma.
🎉Обязательно к прочтению новичкам: полное руководство по написанию мини-программ WeChat с использованием программного обеспечения Cursor.
[Забавный проект Docker] VoceChat — еще одно приложение для мгновенного чата (IM)! Может быть встроен в любую веб-страницу!

Как реализовать переход по странице в HTML (html переходит на указанную страницу)

Как решить проблему зависания и низкой скорости при установке зависимостей с помощью npm. Существуют ли доступные источники npm, которые могут решить эту проблему?

Серия From Zero to Fun: Uni-App WeChat Payment Practice WeChat авторизует вход в систему и украшает страницу заказа, создает интерфейс заказа и инициирует запрос заказа

Серия uni-app: uni.navigateЧтобы передать скачок значения

Апплет WeChat настраивает верхнюю панель навигации и адаптируется к различным моделям.

JS-время конвертации

Обеспечьте бесперебойную работу ChromeDriver 125: советы по решению проблемы chromedriver.exe не найдены

Поле комментария, щелчок мышью, специальные эффекты, js-код
Объект массива перемещения объекта JS

Как открыть разрешение на позиционирование апплета WeChat_Как использовать WeChat для определения местонахождения друзей
Я даю вам два набора из 18 простых в использовании фонов холста Power BI, так что вам больше не придется возиться с цветами!

Получить текущее время в js_Как динамически отображать дату и время в js

Вам необходимо изучить сочетания клавиш vsCode для форматирования и организации кода, чтобы вам больше не приходилось настраивать формат вручную.

У ChatGPT большое обновление. Всего за 45 минут пресс-конференция показывает, что OpenAI сделал еще один шаг вперед.
Copilot облачной разработки — упрощение разработки

Микросборка xChatGPT с низким кодом, создание апплета чат-бота с искусственным интеллектом за пять шагов

CUDA Out of Memory: идеальное решение проблемы нехватки памяти CUDA
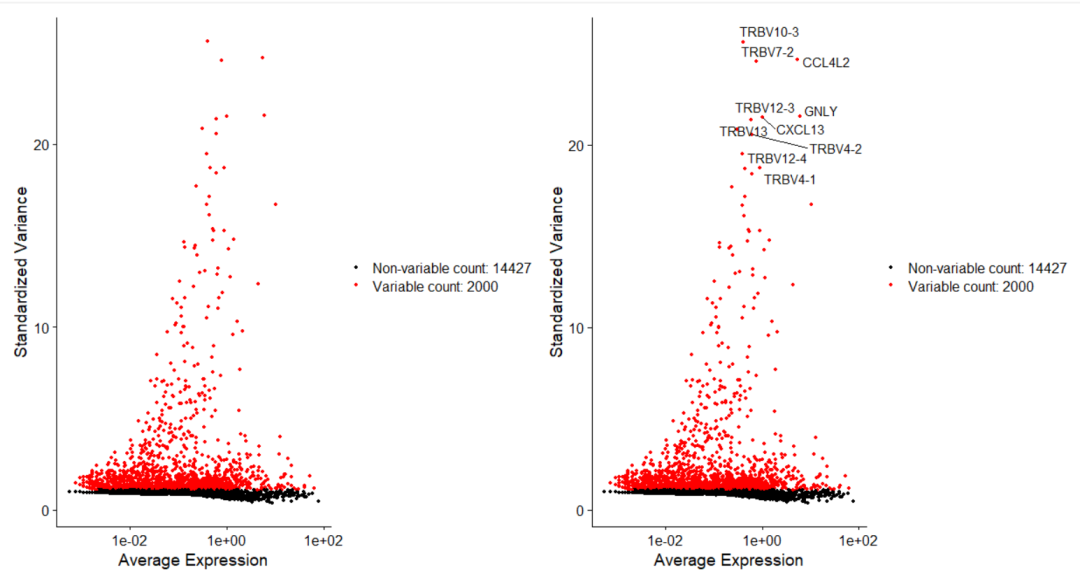
Анализ кластеризации отдельных ячеек, который должен освоить каждый&MarkerгенетическийВизуализация

vLLM: мощный инструмент для ускорения вывода ИИ

CodeGeeX: мощный инструмент генерации кода искусственного интеллекта, который можно использовать бесплатно в дополнение к второму пилоту.

Машинное обучение Реальный бой LightGBM + настройка параметров случайного поиска: точность 96,67%
Бесшовная интеграция, мгновенный интеллект [1]: платформа больших моделей Dify-LLM, интеграция без кодирования и встраивание в сторонние системы, более 42 тысяч звезд, чтобы стать свидетелями эксклюзивных интеллектуальных решений.
LM Studio для создания локальных больших моделей
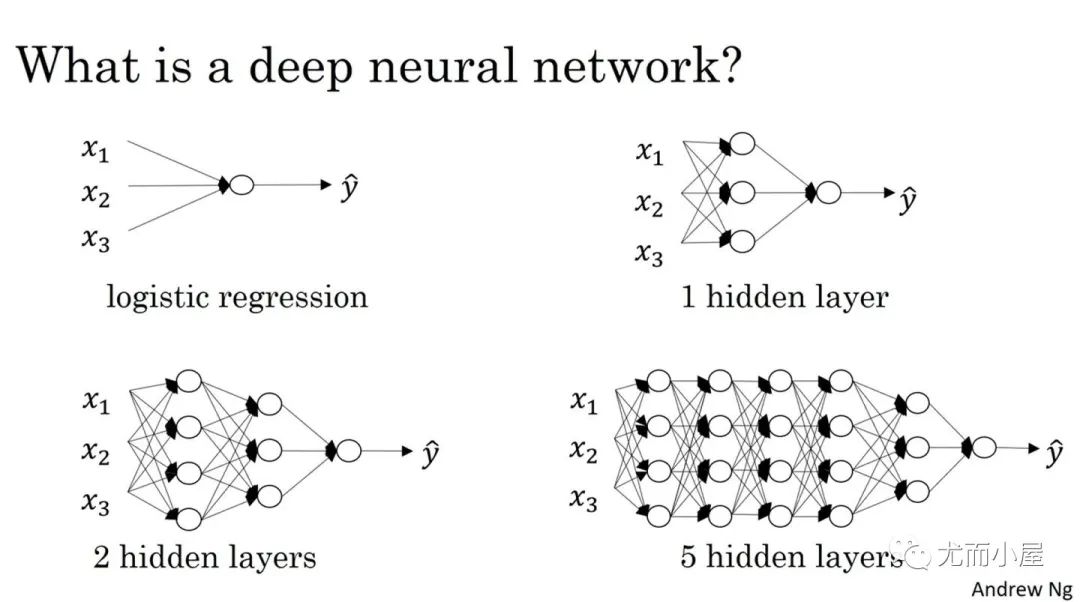
Как определить количество слоев и нейронов скрытых слоев нейронной сети?
