Код всего процесса менделевского промежуточного анализа (1)

фон:Это наблюдается,Более высокий уровень обучения связан с меньшим риском болезни Альцгеймера. Однако,Биологические механизмы, лежащие в основе этой ассоциации, остаются неясными. Защитный эффект обучения против болезни Альцгеймера может быть опосредован увеличением резерва мозга.
метод:Для исследования мы используем двухвыборочную менделевскую рандомизацию.обучатьстепень、Предполагаемая причинно-следственная связь между структурным резервом мозга, представленным фенотипами МРТ, и болезнью Альцгеймера。Мы сделали заказпеременная Менделевский рандомизационный анализ,Изучить двунаправленную связь между (i) степенью обучения и болезнью Альцгеймера; (ii) степенью обучения и фенотипом визуализации и (iii) фенотипом визуализации и болезнью Альцгеймера; Множественная переменная менделевская рандомизация использовалась для оценки того, опосредуют ли структурные фенотипы мозга влияние обучения на риск болезни Альцгеймера.
результат:нашрезультатподдерживатьобучатьстепень对阿尔茨海默病风险的保护性因果效应,и потенциальные двунаправленные причинно-следственные связи между степенью обученности и макро- и микроструктурой мозга. Однако,Мы не нашли доказательств того, что эти структурные маркеры влияют на риск болезни Альцгеймера. обучать Степень защиты от болезни Альцгеймера может быть достигнута за счет других мер резерва мозга, не включенных в это исследование.,или опосредовано другими механизмами.
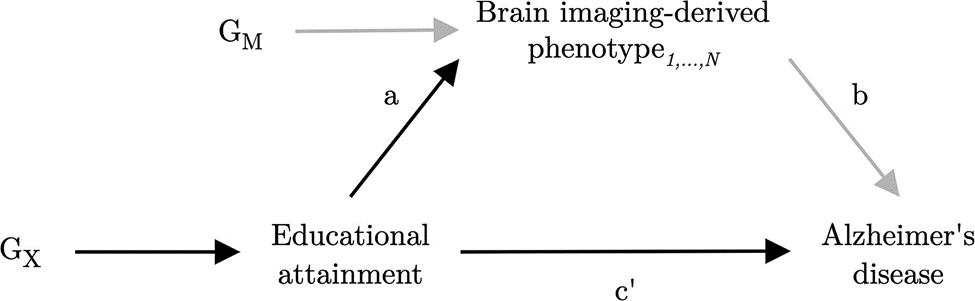
GX представляет собой набор инструментальных переменных советника. Параметр a представляет собой прямое причинное влияние ЭА на фенотипы 1, ..., N, полученные при визуализации, а параметр b представляет прямое причинное влияние фенотипов 1, ..., N, полученных при визуализации, на болезнь Альцгеймера. Параметр c' представляет собой прямое причинное влияние ЭА на болезнь Альцгеймера. Параметры a и c' оцениваются с использованием набора переменных GX, а параметр b оценивается с использованием набора переменных GM. Смешивающие переменные не показаны на рисунке.
Примерно уяснив идею статьи, давайте посмотрим, есть ли что-то, что нас больше всего беспокоит——
R code for reproducing all MR analyses can be found on https://github.com/as2970/EA_brain_AD_MR.
чудесный! ! ! Спасибо автору статьи~
основной код шага 0
library(tidyverse)
library(data.table)
# library(ivpack)
library(meta)
library(devtools)
library(pacman)
library(TwoSampleMR)
library(MRInstruments)
library(ieugwasr)
# install_github("phenoscanner/phenoscanner")
library(phenoscanner)
library(LDlinkR)
library(mr.raps)
library(MRPRESSO)
library(extrafont)
library(anchors)
Здесь автор написал функцию, если вы когда-нибудь
Введение в биокредит&данные Откройте для себя онлайн-классы в прямом эфире6月班
Блок интеллектуального анализа данных GEO, прошедший обучение на марафоне
Если у вас есть определенные знания языка R, вы можете попытаться понять эту функцию. Последующий анализ будет значительно упрощен⬇.
## MR_prep <- function(exp, out) {
# Extracting instruments
exp_dat <- read_exposure_data(
filename = paste0(deparse(substitute(exp)), "_exposure.txt", sep = ""),
clump = FALSE,
sep = " ",
snp_col = "SNP",
beta_col = "BETA",
se_col = "SE",
eaf_col = "AF1",
effect_allele_col = "A1",
other_allele_col = "A2",
pval_col = "P",
ncase_col = "N_case",
ncontrol_col = "N_control",
samplesize_col = "N",
min_pval = 1e-200,
log_pval = FALSE,
chr_col = "CHR",
pos_col = "POS"
)
# Clumping instruments
exp_dat <- exp_dat %>%
rename(
rsid = SNP,
pval = pval.exposure
)
exp_dat_clumped <- ld_clump(
dat = exp_dat,
clump_kb = 10000,
clump_r2 = 0.001,
clump_p = 5e-8,
plink_bin = genetics.binaRies::get_plink_binary(),
bfile = "" #path to LD reference dataset
)
exp_dat_clumped <- exp_dat_clumped %>%
rename(
SNP = rsid,
pval.exposure = pval
)
# Printing number of IVs for exposure
print(paste0("Number of IVs: ", as.character(length(exp_dat_clumped$SNP))))
Чтение конечных данных
# Extracting instruments from outcome GWAS
out_dat <- read_outcome_data(
filename = paste0(deparse(substitute(out)), "_outcome.txt", sep = ""),
snps = exp_dat_clumped$SNP,
sep = " ",
snp_col = "SNP",
beta_col = "BETA",
se_col = "SE",
eaf_col = "AF1",
effect_allele_col = "A1",
other_allele_col = "A2",
pval_col = "P",
ncase_col = "N_case",
ncontrol_col = "N_control",
samplesize_col = "N",
min_pval = 1e-200,
log_pval = FALSE,
chr_col = "CHR",
pos_col = "POS"
)
# Identifying & printing exposure instruments missing from outcome GWAS
missing_IVs <- exp_dat_clumped$SNP[!(exp_dat_clumped$SNP %in% out_dat$SNP)]
print(paste0("Number of IVs missing from outcome GWAS: ", as.character(length(missing_IVs))))
print("List of IVs missing from outcome GWAS:")
for (i in 1:length(missing_IVs)) {
print(paste0(missing_IVs[i]))
}
Поиск прокси-SNP
# Replacing missing instruments from outcome GWAS with proxies
if(length(missing_IVs) == 0) {
print("All exposure IVs found in outcome GWAS.")
} else {
print("Some exposure IVs missing from outcome GWAS.")
out_full <- fread(paste0(deparse(substitute(out)), "_outcome.txt", sep = ""))
for (i in 1:length(missing_IVs)) {
proxies <- LDproxy(snp = missing_IVs[i], pop = "EUR", r2d = "r2", token = "6fb632e022ef", file = FALSE)
proxies <- proxies[proxies$R2 > 0.8, ]
proxy_present = FALSE
if(length(proxies$RS_Number) == 0){
print(paste0("No proxy SNP available for ", missing_IVs[i]))
} else {
for (j in 1:length(proxies$RS_Number)) {
proxy_present <- proxies$RS_Number[j] %in% out_full$SNP
if (proxy_present) {
proxy_SNP = proxies$RS_Number[j]
proxy_SNP_allele_1 = str_sub(proxies$Alleles[j], 2, 2)
proxy_SNP_allele_2 = str_sub(proxies$Alleles[j], 4, 4)
original_SNP_allele_1 = str_sub(proxies$Alleles[1], 2, 2)
original_SNP_allele_2 = str_sub(proxies$Alleles[1], 4, 4)
break
}
}
}
if(proxy_present == TRUE) {
print(paste0("Proxy SNP found. ", missing_IVs[i], " replaced with ", proxy_SNP))
proxy_row <- out_dat[1, ]
proxy_row$SNP = missing_IVs[i]
proxy_row$beta.outcome = as.numeric(out_full[out_full$SNP == proxy_SNP, "BETA"])
proxy_row$se.outcome = as.numeric(out_full[out_full$SNP == proxy_SNP, "SE"])
if (out_full[out_full$SNP == proxy_SNP, "A1"] == proxy_SNP_allele_1) proxy_row$effect_allele.outcome = original_SNP_allele_1
if (out_full[out_full$SNP == proxy_SNP, "A1"] == proxy_SNP_allele_2) proxy_row$effect_allele.outcome = original_SNP_allele_2
if (out_full[out_full$SNP == proxy_SNP, "A2"] == proxy_SNP_allele_1) proxy_row$other_allele.outcome = original_SNP_allele_1
if (out_full[out_full$SNP == proxy_SNP, "A2"] == proxy_SNP_allele_2) proxy_row$other_allele.outcome = original_SNP_allele_2
proxy_row$pval.outcome = as.numeric(out_full[out_full$SNP == proxy_SNP, "P"])
proxy_row$samplesize.outcome = as.numeric(out_full[out_full$SNP == proxy_SNP, "N"])
if("N_case" %in% colnames(out_full)) proxy_row$ncase.outcome = as.numeric(out_full[out_full$SNP == proxy_SNP, "N_case"])
if("N_control" %in% colnames(out_full))proxy_row$ncontrol.outcome = as.numeric(out_full[out_full$SNP == proxy_SNP, "N_control"])
proxy_row$chr.outcome = as.numeric(exp_dat_clumped[exp_dat_clumped$SNP == missing_IVs[i], "chr.exposure"])
proxy_row$pos.outcome = as.numeric(exp_dat_clumped[exp_dat_clumped$SNP == missing_IVs[i], "pos.exposure"])
if("AF1" %in% colnames(out_full)) proxy_row$eaf.outcome = as.numeric(out_full[out_full$SNP == proxy_SNP, "AF1"])
out_dat <- rbind(out_dat, proxy_row)
}
if(proxy_present == FALSE) {
print(paste0("No proxy SNP available for ", missing_IVs[i], " in outcome GWAS."))
}
}
}
затем гармонизировать
# Harmonising exposure and outcome datasets
dat <- harmonise_data(
exposure_dat = exp_dat_clumped,
outcome_dat = out_dat,
action = 2
)
## } ##Конец функции самостоятельного написания
Эта функция очень длинная,Поначалу может показаться немного трудным смотреть,Не волнуйтесь,Используйте проблемы с пользой??функция`,Все желающие также могут обсудить и решить проблему в комментариях~
напримерread_exposure_dataфункция,Когда мы готовимся предоставить данные, мы должны учитывать тип данных, требуемых этой функцией.,Вот и все??read_exposure_dataВзгляните,В основном смотримArguments,Что необходимо,Какие данные могут оказаться ненужными.

??read_outcome_dataфункция То же самое верно,Это то, что нам нужно знать заранее, на этапе подготовки! ! ! Все нужно делать заранее (Урок редактора...)
Советы
Что касается конвертации eaf и maf, то это небольшая деталь, но не стоит быть небрежным~
Поместите это здесь для справки
【https://www.bilibili.com/video/BV1De4y1577E/?vd_source=e5e36ce10569a7a5d647d18bdb42e4b5】
for (i in 1:length(BRCA1_TN$eaf_col)) {
if (BRCA1_TN$eaf_col[i]<0.5) {BRCA1_TN$eaf_col[i]= 1 -BRCA1_TN$eaf_col[i]
}else{BRCA1_TN$eaf_col[i] = BRCA1_TN$eaf_col[i]}
}
Нужно выполнить 4 шага кода, давайте идти шаг за шагом~


Учебное пособие по Jetpack Compose для начинающих, базовые элементы управления и макет

Код js веб-страницы, фон частицы, код спецэффектов

【новый! Суперподробное】Полное руководство по свойствам компонентов Figma.
🎉Обязательно к прочтению новичкам: полное руководство по написанию мини-программ WeChat с использованием программного обеспечения Cursor.
[Забавный проект Docker] VoceChat — еще одно приложение для мгновенного чата (IM)! Может быть встроен в любую веб-страницу!

Как реализовать переход по странице в HTML (html переходит на указанную страницу)

Как решить проблему зависания и низкой скорости при установке зависимостей с помощью npm. Существуют ли доступные источники npm, которые могут решить эту проблему?

Серия From Zero to Fun: Uni-App WeChat Payment Practice WeChat авторизует вход в систему и украшает страницу заказа, создает интерфейс заказа и инициирует запрос заказа

Серия uni-app: uni.navigateЧтобы передать скачок значения

Апплет WeChat настраивает верхнюю панель навигации и адаптируется к различным моделям.

JS-время конвертации

Обеспечьте бесперебойную работу ChromeDriver 125: советы по решению проблемы chromedriver.exe не найдены

Поле комментария, щелчок мышью, специальные эффекты, js-код
Объект массива перемещения объекта JS

Как открыть разрешение на позиционирование апплета WeChat_Как использовать WeChat для определения местонахождения друзей
Я даю вам два набора из 18 простых в использовании фонов холста Power BI, так что вам больше не придется возиться с цветами!

Получить текущее время в js_Как динамически отображать дату и время в js

Вам необходимо изучить сочетания клавиш vsCode для форматирования и организации кода, чтобы вам больше не приходилось настраивать формат вручную.

У ChatGPT большое обновление. Всего за 45 минут пресс-конференция показывает, что OpenAI сделал еще один шаг вперед.
Copilot облачной разработки — упрощение разработки

Микросборка xChatGPT с низким кодом, создание апплета чат-бота с искусственным интеллектом за пять шагов

CUDA Out of Memory: идеальное решение проблемы нехватки памяти CUDA
Анализ кластеризации отдельных ячеек, который должен освоить каждый&MarkerгенетическийВизуализация

vLLM: мощный инструмент для ускорения вывода ИИ

CodeGeeX: мощный инструмент генерации кода искусственного интеллекта, который можно использовать бесплатно в дополнение к второму пилоту.

Машинное обучение Реальный бой LightGBM + настройка параметров случайного поиска: точность 96,67%
Бесшовная интеграция, мгновенный интеллект [1]: платформа больших моделей Dify-LLM, интеграция без кодирования и встраивание в сторонние системы, более 42 тысяч звезд, чтобы стать свидетелями эксклюзивных интеллектуальных решений.
LM Studio для создания локальных больших моделей
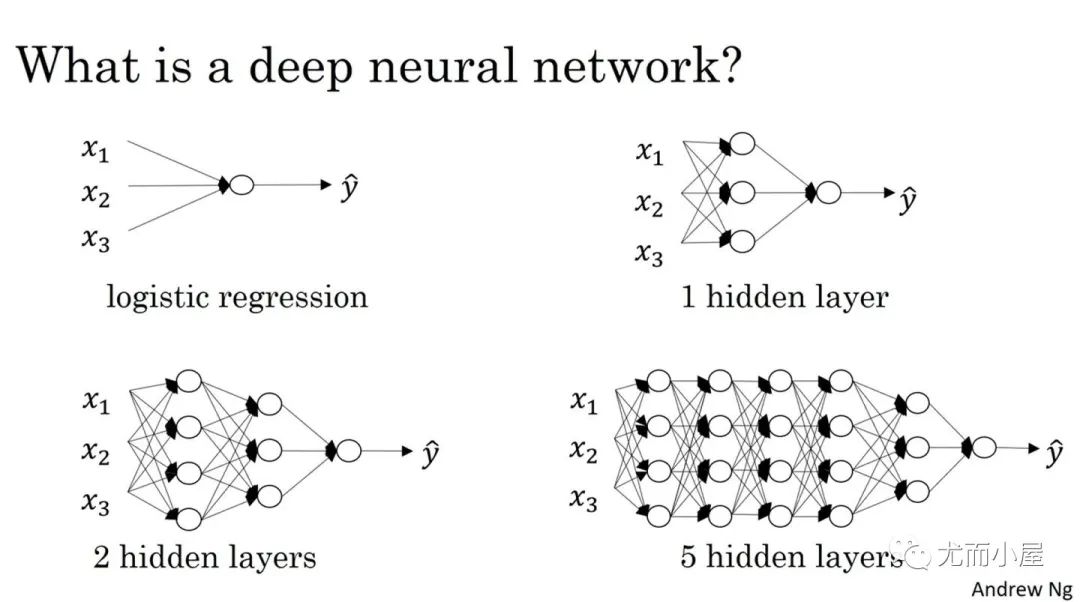
Как определить количество слоев и нейронов скрытых слоев нейронной сети?
