Используйте Seurat v5 для чтения нескольких 10-кратных матриц транскриптома одной клетки
Хотя сказано, что это несколько семплов, автор сгруппировал их в трехфайловый формат с 10 сэмплами. Так что читать легко. Далее мы демонстрируем настоящий Seurat v5 для чтения нескольких 10-кратных матриц одноклеточного транскриптома. Набор данных находится в https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE162616 Вы можете увидеть Матрицу, предоставленная автором Весьма3 стандартных файла для файлов 10X,Но под каждым образцом лежит 3 файла,Вам просто нужно изменить имя файла соответствующим образом:
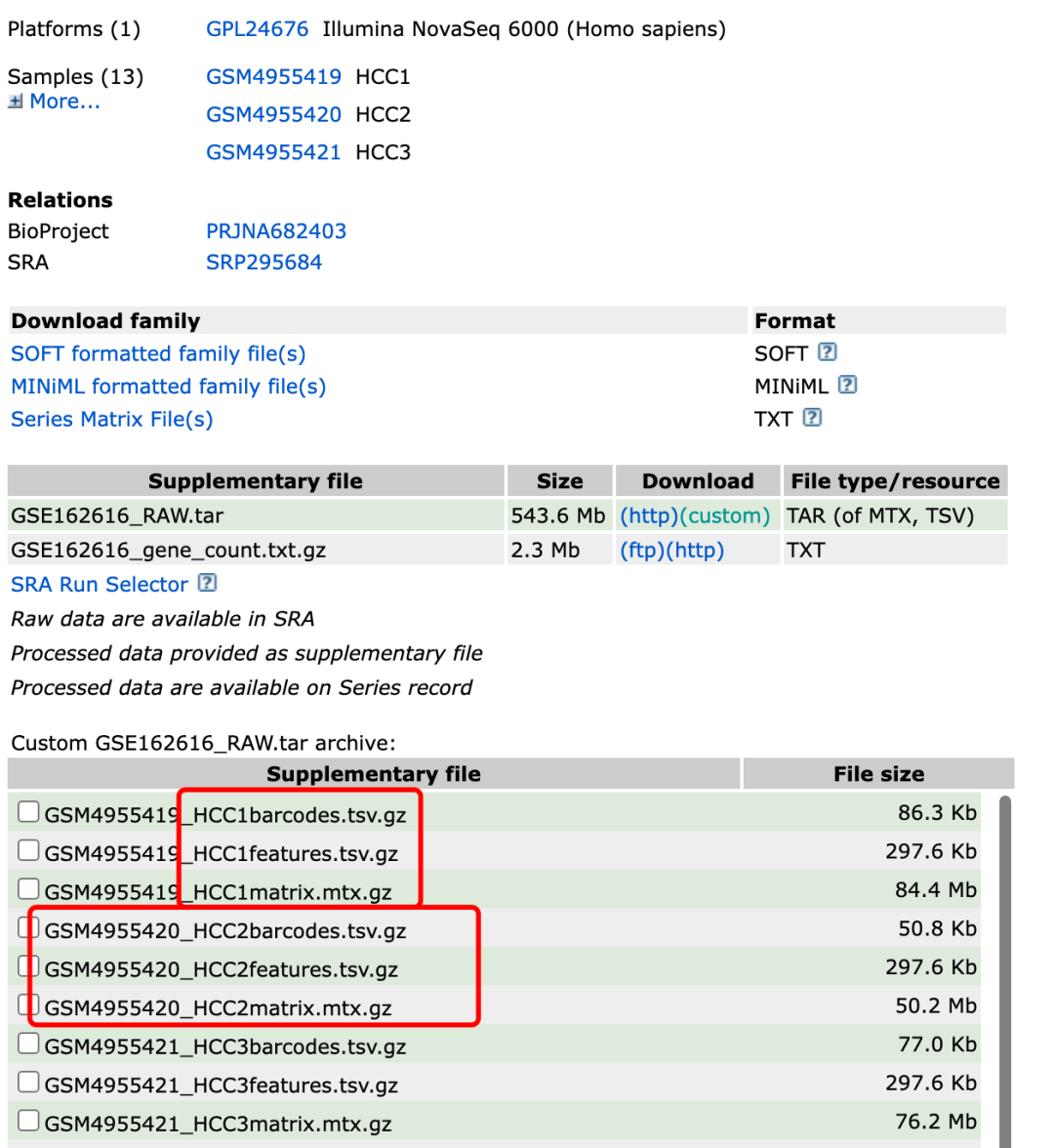
Матрица, предоставленная автором
После нашей модификации у каждого образца есть папка,Внутри каждой папки находится3 стандартных файла для файлов 10X, как показано ниже:
$ tree -h GSE162616_RAW/outputs/
GSE162616_RAW/outputs/
|-- [ 0] HCC1
| |-- [ 86K] barcodes.tsv.gz
| |-- [298K] features.tsv.gz
| `-- [ 84M] matrix.mtx.gz
|-- [ 0] HCC2
| |-- [ 51K] barcodes.tsv.gz
| |-- [298K] features.tsv.gz
| `-- [ 50M] matrix.mtx.gz
`-- [ 0] HCC3
|-- [ 77K] barcodes.tsv.gz
|-- [298K] features.tsv.gz
`-- [ 76M] matrix.mtx.gz
3 directories, 9 files
Если читать по предыдущей версии Seurat V4, код будет следующим:
dir='GSE162616_RAW/outputs/'
samples=list.files( dir )
samples
library(data.table)
sceList = lapply(samples,function(pro){
# pro=samples[1]
print(pro)
sce=CreateSeuratObject(counts = Read10X(file.path(dir,pro) ) ,
project = pro ,
min.cells = 5,
min.features = 300,)
return(sce)
})
names(sceList)
library(stringr)
# samples=gsub('.txt.gz','',str_split(samples,'_',simplify = T)[,2])
samples
names(sceList) = samples
sce.all <- merge(sceList[[1]], y= sceList[ -1 ] ,
add.cell.ids = samples)
as.data.frame(sce.all@assays$RNA$counts[1:10, 1:2])
# only the first layer is used
head(sce.all@meta.data, 10)
table(sce.all@meta.data$orig.ident)
Я писал приведенный выше код в течение многих лет, и он не обновлялся и не улучшался. Мы полагаемся на эту версию процесса Сёра V4 для создания большого количества процессов кластеризации уменьшения размерности транскриптома одной клетки для общедоступных наборов данных, более чем. 100 Вся обработка общедоступных наборов данных одной ячейки, ссылка: https://pan.baidu.com/s/1MzfqW07P9ZqEA_URQ6rLbA?pwd=3heo Но недавно его официальная версия стала V5...
Поскольку это версия Seurat V5, если несколько файлов считываются отдельно, функция слияния фактически не объединяет матрицу выражений каждого образца, как показано ниже:
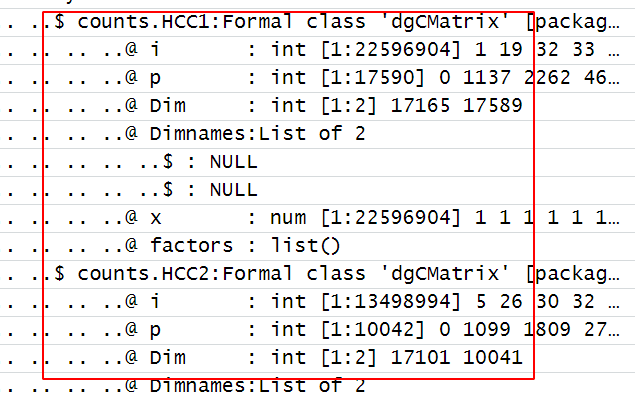
Как видите, каждая выборка в объекте Сёра по-прежнему представляет собой независимую матрицу. . . .
В этом случае последующий процесс не будет продолжен. На данный момент у нас есть очень простой способ избежать слияния после отдельного чтения, как показано ниже:
tmp = list.dirs('GSE162616_RAW/outputs/')[-1]
tmp
ct = Read10X(tmp)
sce.all=CreateSeuratObject(counts = ct ,
min.cells = 5,
min.features = 300,)
Фактически, это происходит потому, что эта функция Read10X может считывать несколько разумных путей одновременно, поэтому наши три примера, показанные ниже, равномерно считываются в разреженную матрицу вместо независимой разреженной матрицы для каждого образца, как показано ниже:
> tmp
[1] "GSE162616_RAW/outputs/HCC1"
[2] "GSE162616_RAW/outputs/HCC2"
[3] "GSE162616_RAW/outputs/HCC3"
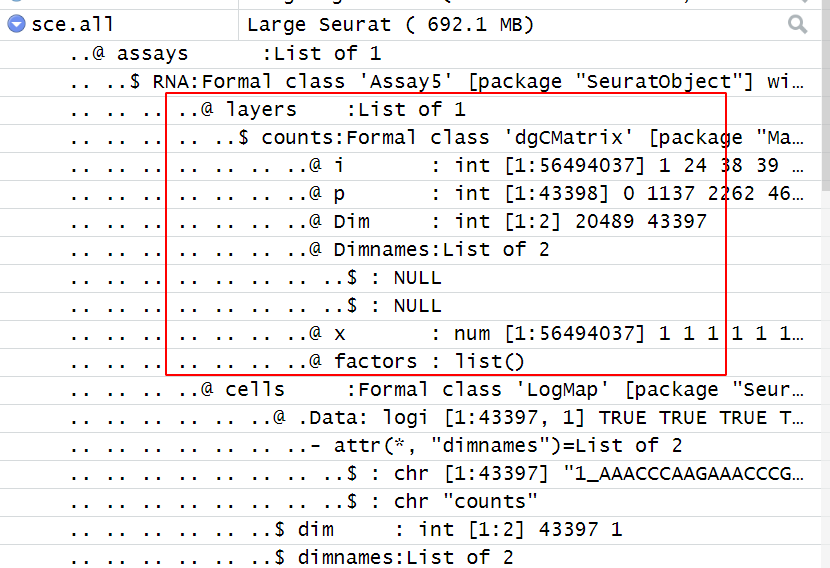
Единое чтение становится разреженной матрицей
Если структура функции или объекта Сёра не ясна, произойдет следующий неправильный метод чтения:
> sce.all=CreateSeuratObject(counts = Read10X('GSE162616_RAW/outputs/') ,
+ min.cells = 5,
+ min.features = 300,)
Error in Read10X("GSE162616_RAW/outputs/") :
Barcode file missing. Expecting barcodes.tsv.gz
эта функция Read10X может принимать один или несколько разумных путей.,Разумный путь — сказать, что существует3 стандартных файла для файлов 10X,Разве это не очень просто?
Позже мы также продемонстрируем, как читать несколько образцов одноклеточного транскриптома, но матрицы этих образцов не имеют файлового формата 10x3, поэтому это будет сложнее!



Углубленный анализ переполнения памяти CUDA: OutOfMemoryError: CUDA не хватает памяти. Попыталась выделить 3,21 Ги Б (GPU 0; всего 8,00 Ги Б).
[Решено] ошибка установки conda. Среда решения: не удалось выполнить первоначальное зависание. Повторная попытка с помощью файла (графическое руководство).
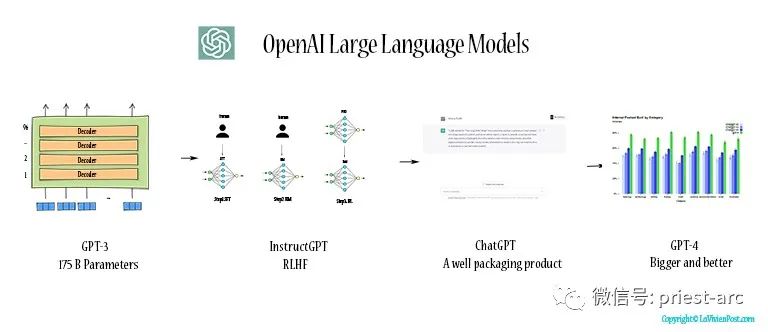
Прочитайте нейросетевую модель Трансформера в одной статье

.ART Теплые зимние предложения уже открыты

Сравнительная таблица описания кодов ошибок Amap

Уведомление о последних правилах Points Mall в декабре 2022 года.

Даже новички могут быстро приступить к работе с легким сервером приложений.
Взгляд на RSAC 2024|Защита конфиденциальности в эпоху больших моделей
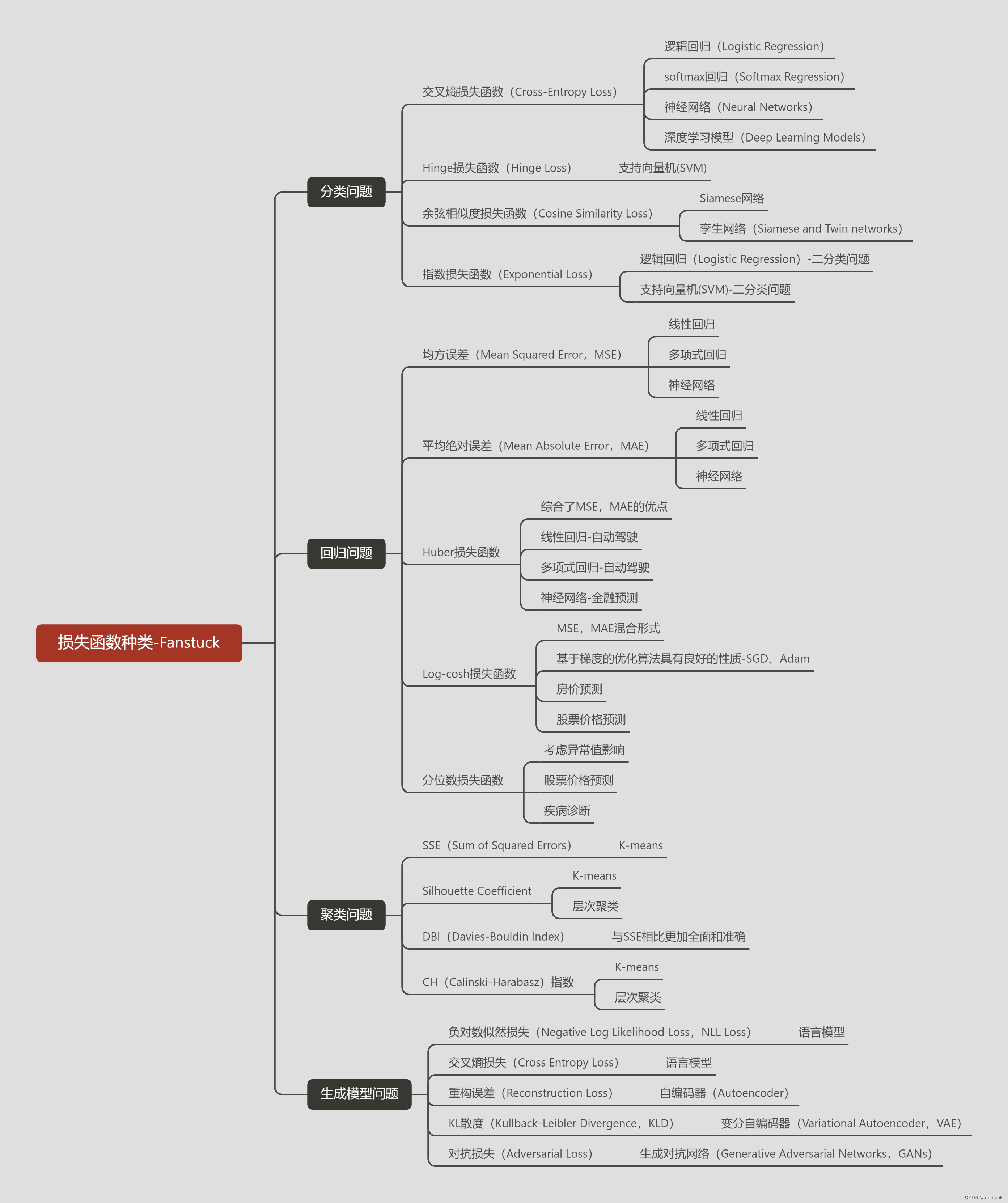
Вы используете ИИ каждый день и до сих пор не знаете, как ИИ дает обратную связь? Одна статья для понимания реализации в коде Python общих функций потерь генеративных моделей + анализ принципов расчета.

Используйте (внутренний) почтовый ящик для образовательных учреждений, чтобы использовать Microsoft Family Bucket (1T дискового пространства на одном диске и версию Office 365 для образовательных учреждений)
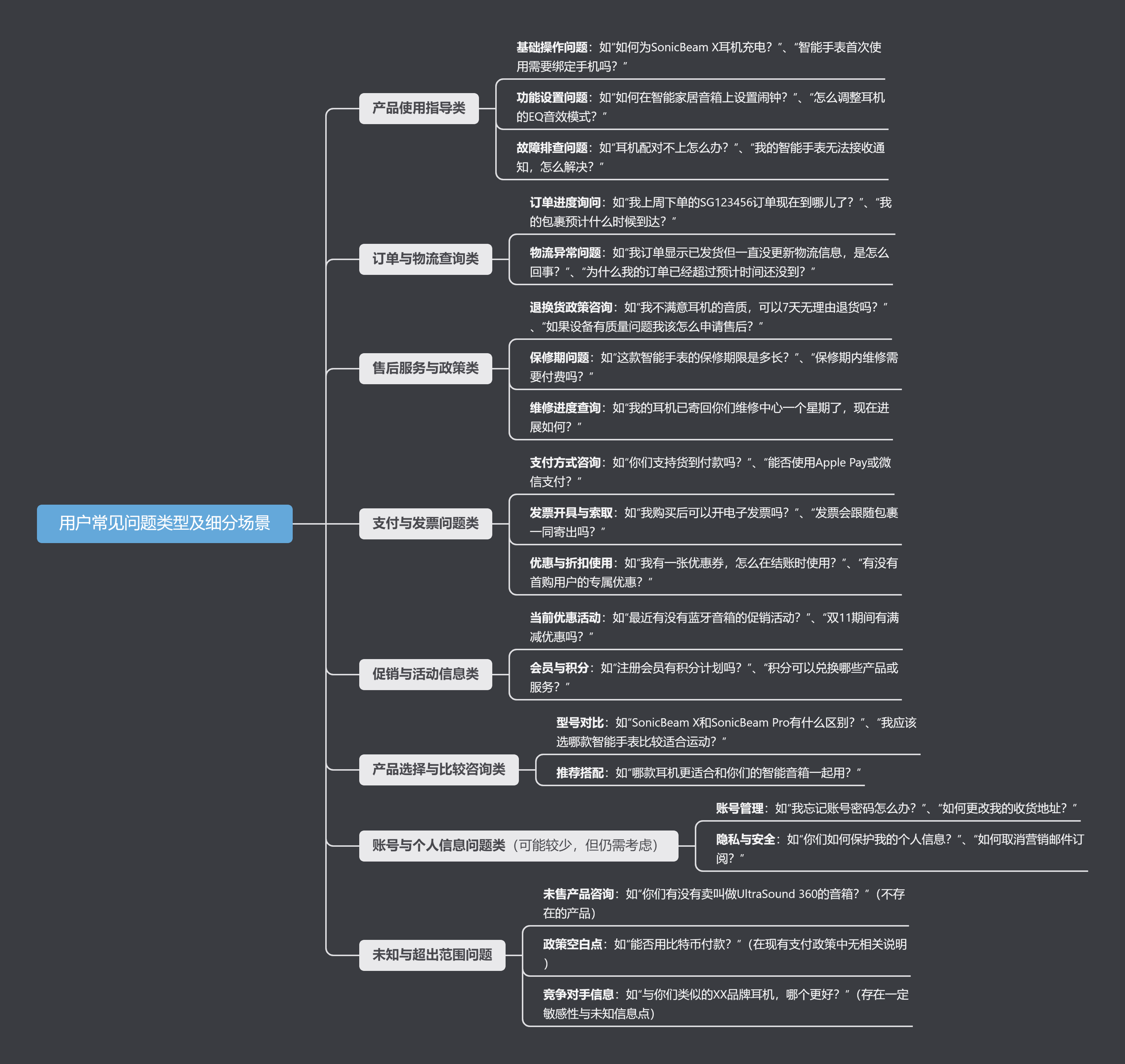
Руководство по началу работы с оперативным проектом (7) Практическое сочетание оперативного письма — оперативного письма на основе интеллектуальной системы вопросов и ответов службы поддержки клиентов

[docker] Версия сервера «Чтение 3» — создайте свою собственную программу чтения веб-текста

Обзор Cloud-init и этапы создания в рамках PVE
Корпоративные пользователи используют пакет регистрационных ресурсов для регистрации ICP для веб-сайта и активации оплаты WeChat H5 (с кодом платежного узла версии API V3)

Подробное объяснение таких показателей производительности с высоким уровнем параллелизма, как QPS, TPS, RT и пропускная способность.
Удачи в конкурсе Python Essay Challenge, станьте первым, кто испытает новую функцию сообщества [Запускать блоки кода онлайн] и выиграйте множество изысканных подарков!

[Техническая посадка травы] Кровавая рвота и отделка позволяют вам необычным образом ощипывать гусиные перья! Не распространяйте информацию! ! !

[Официальное ограниченное по времени мероприятие] Сейчас ноябрь, напишите и получите приз

Прочтите это в одной статье: Учебник для няни по созданию сервера Huanshou Parlu на базе CVM-сервера.

Cloud Native | Что такое CRD (настраиваемые определения ресурсов) в K8s?

Как использовать Cloudflare CDN для настройки узла (CF самостоятельно выбирает IP) Гонконг, Китай/Азия узел/сводка и рекомендации внутреннего высокоскоростного IP-сегмента

Дополнительные правила вознаграждения амбассадоров акции в марте 2023 г.
Можно ли открыть частный сервер Phantom Beast Palu одним щелчком мыши? Супер простой урок для начинающих! (Прилагается метод обновления сервера)
[Играйте с Phantom Beast Palu] Обновите игровой сервер Phantom Beast Pallu одним щелчком мыши

Maotouhu делится: последний доступный внутри страны адрес склада исходного образа Docker 2024 года (обновлено 1 декабря)

Кодирование Base64 в MultipartFile
5 точек расширения SpringBoot, супер практично!

Глубокое понимание сопоставления индексов Elasticsearch.
15 рекомендуемых платформ разработки с нулевым кодом корпоративного уровня. Всегда найдется та, которая вам понравится.
