Фильтрация с помощью ncount_RNA и nFeature_RNA
Предварительное резюме
В прошлый раз я вкратце это для вас организовал.Понимание кривых идентификации клеток,вИспользуйте nCount_RNA или nFeature_RNA, чтобы нарисовать кривую идентификации клеток на языке R и найти подходящее пороговое значение.,Был проведен предварительный контроль качества.
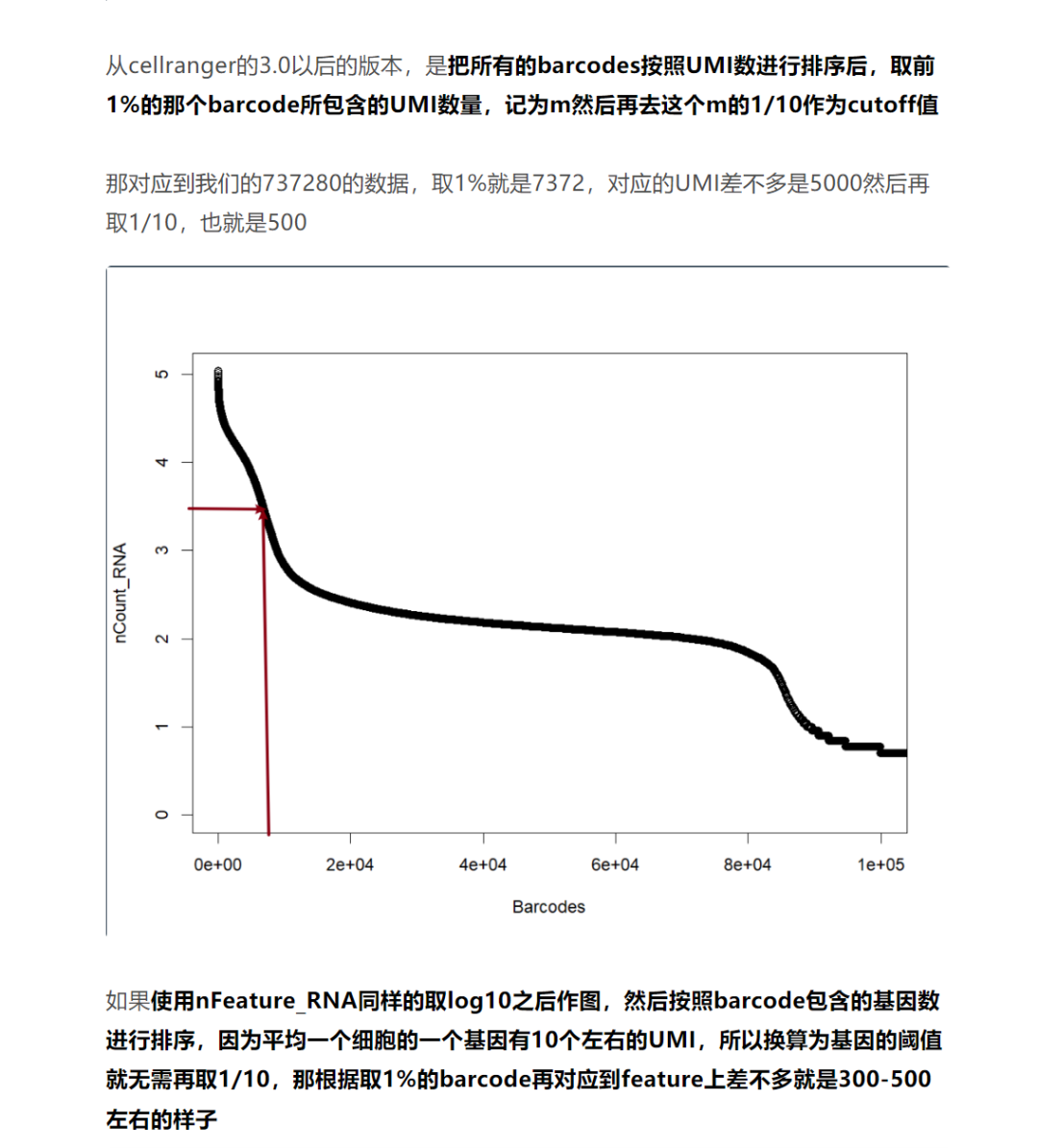
Также упоминается в конце,Редко в дальнейшем будут исходные необработанные данные.,Обычно мы используемcellrangerПосле контроля качестваданныепровести анализ。Однако для обработанныхданныенабор, мы можемВизуализируйте nFeature_RNA и nCount_RNA, чтобы помочь в контроле качества.
Тогда сначала мына основеУчебники с официального сайта SeuratРазберемся и рассмотримnFeature_RNA и nCount_RNA,И предварительный просмотр определяет порог,Затем узнайте о применении в реальных ситуациях анализа.。
Введение в nFeature_RNA и nCount_RNA
После создания объекта seurat,когда ничего не делаешь,seurat создаст метаданные для каждой ячейки и сохранит их в файле метаданные.
#readданныеcreate seuratobject
pbmc.data <- Read10X(data.dir = "./filtered_gene_bc_matrices/hg19/")
pbmc <- CreateSeuratObject(counts = pbmc.data,
project = "pbmc3k",
min.cells = 3)
> dim(pbmc)
[1] 13714 2700

Содержание каждого столбца:
orig.ident:Обычно содержит известное название образца,По умолчанию используется значение, которое мы присвоили проекту.,Если значение не присвоено, это SeuratProject.nCount_RNA:каждой ячейкиUMIчислоnFeature_RNA:обнаруживается в каждой ячейкеиз Генчисло
можно увидетьnCount_RNAиnFeature_RNAЕсть еще различия,Это связано с ихМетод расчетасвязанный
#nCount_RNA: общее количество UMI — это количество транскриптов.
colSums(sce@assays$RNA$counts)
#nFeature_RNA: общее количество генов
colSums(sce@assays$RNA$counts>0)
Визуализация и пороговое суждение
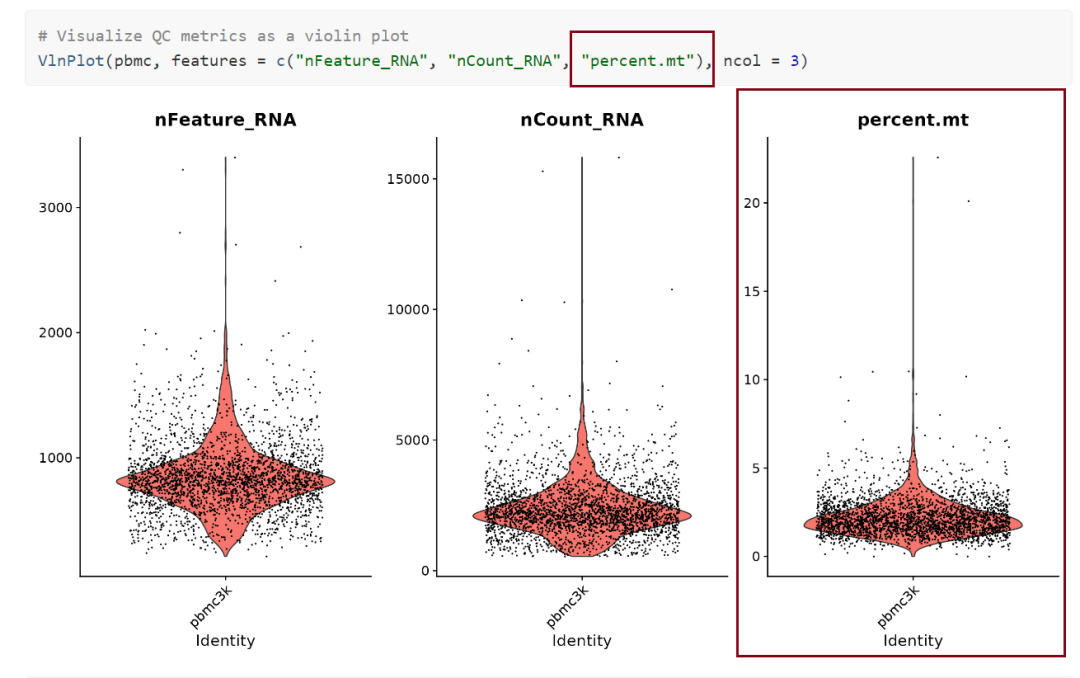
Вы можете использовать графики скрипки, чтобы просто визуализировать nFeature_RNA и nCount_RNA.
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA"))
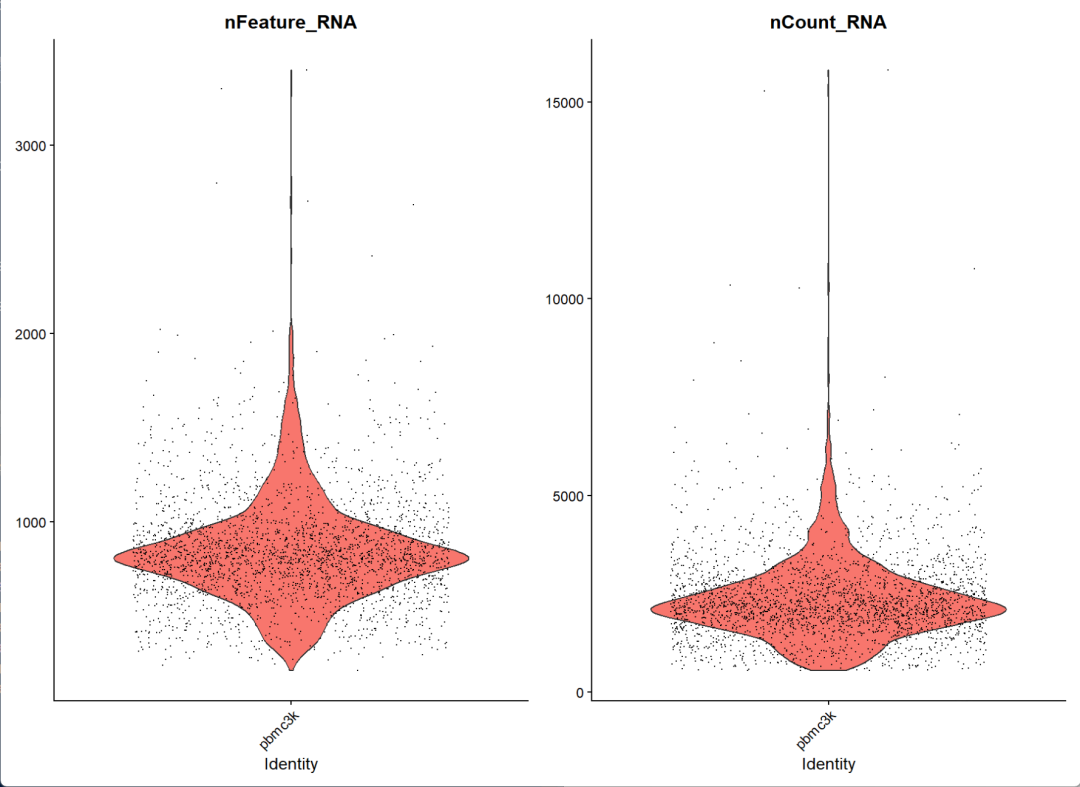
Перед фильтрацией
диаграмма nFeature_RNA:Отражает количество генов, экспрессируемых каждой клеткой образца.,Сверхэкспрессия может указывать на двуклеточность или многоклеточность.,Если экспрессия слишком низкая, капли могут быть пустыми или содержать РНК из окружающей среды.
график nCount_RNA:отражает содержимое, содержащееся в каждой ячейкеUMIКоличество – это количество транскриптов.
В процессе анализа данных секвенирования 10X Genomics после дегенерации результатов секвенирования через UMI вы можете увидеть, сколько генов было прочитано в клетке. Обычно клетка может получить 40 000–80 000 эффективных UMI. В среднем один ген в клетке имеет около 10 UMI.
Итак, мы делаемпороговое суждениекогда,может быть непосредственно основано наЗначение nFeature_RNA — это количество генов.
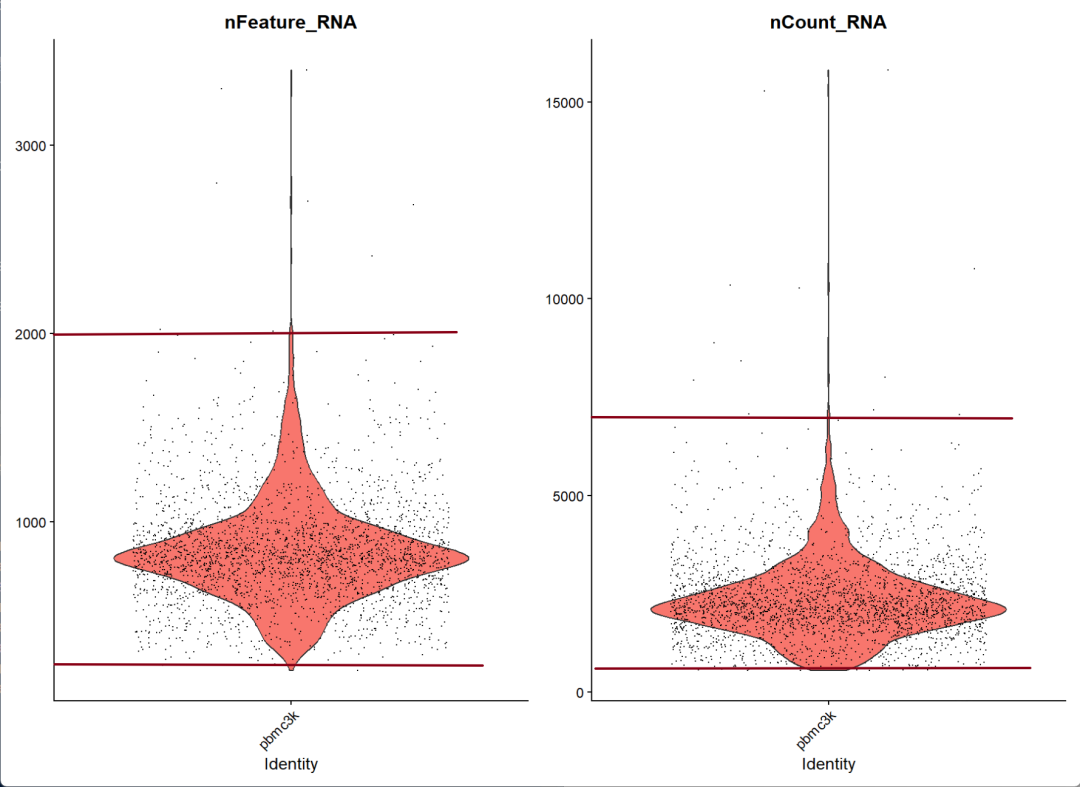
пороговое суждение
We filter cells that have unique feature counts over 2,500 or less than 200
Официальный сайт дает значения больше 200 и меньше 2500, но после визуализации мы видим, что верхний предел равен 2000, что на самом деле нормально.
Однако pbmc — это относительно ранние данные. Количество измеренных ячеек относительно невелико, и верхний предел установлен относительно низко. Если это текущие данные по одной ячейке, все равно требуется специальный анализ данных.
#Фильтрация по пороговому значению и предварительному просмотру
pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2000)
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA"))
> dim(pbmc)
[1] 13714 2692
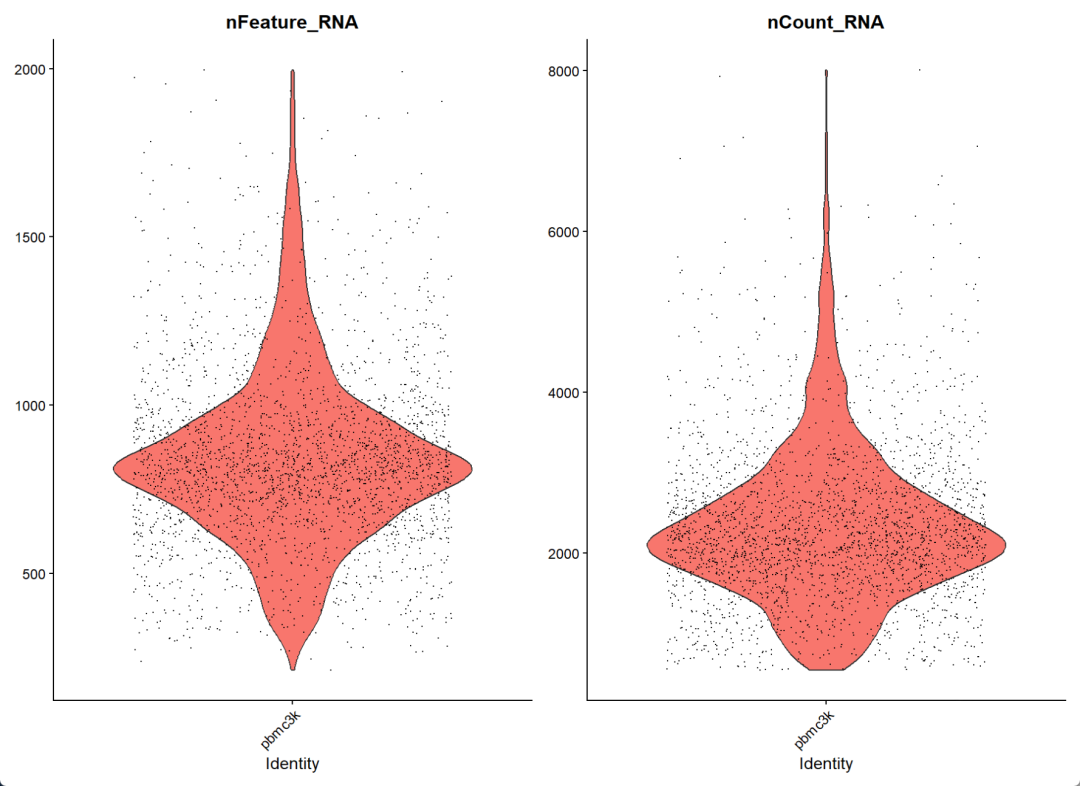
После фильтрации
После фильтрации,Количество ячеек изменилось с исходных 2700 на текущие 2692, а некоторые ячейки были отфильтрованы.
ВышеупомянутоеЧасть контента QC в pbmc3k_tutorial на официальном сайте seurat Далее посмотрим на его применение на реальных данных.
Применение в практическом анализе
Если у вас есть стандартный код анализа для анализа отдельных ячеек в дереве навыков, при необходимости вы можете получить ссылку: https://pan.baidu.com/s/1bIBG9RciAzDhkTKKA7hEfQ?pwd=y4eh
Это в нашемscRNA_scriptsСуществуетqc.Rконтроль качества Скриптдокумент,Он предназначен для контроля качества считываемых данных.
Скрипт Функция сначала вычисляетсяВизуализируется соотношение митохондрий, рибосом и эритроцитов (подробности будут в следующем выпуске), а затем визуализируется состояние этих параметров в клетках.Давайте сначала сосредоточимся на этомnFeature_RNA и nCount_RNA
#qc.RСкриптсерединаnFeature_RNA и Часть nCount_RNA
feats <- c("nFeature_RNA", "nCount_RNA")
p1=VlnPlot(input_sce, group.by = "orig.ident", features = feats, pt.size = 0, ncol = 2) +
NoLegend()
p1
w=length(unique(input_sce$orig.ident))/3+5;w
ggsave(filename="Vlnplot1.pdf",plot=p1,width = w,height = 5)

Перед контролем качества
Как правило, при выполнении стандартного процесса при создании объекта seurat он будет основан на min.cells. = 5 и мин.функции = 300 для фильтрации, поэтому этот этап фильтрации не выполняется в qcСкрипт. Но просто взглянем на Перед фильтрациейизмениться после,мы можемВыполните простую операцию фильтрации на основе результатов визуализации.。

#Простая фильтрация
if(T){
selected_c <- WhichCells(input_sce, expression = nFeature_RNA > 500 & nFeature_RNA < 2500)
selected_f <- rownames(input_sce)[Matrix::rowSums(input_sce@assays$RNA$counts > 0 ) > 3]
input_sce.filt <- subset(input_sce, features = selected_f, cells = selected_c)
dim(input_sce)
dim(input_sce.filt)
}
#Визуализация После фильтрациииз Состояние feats <- c("nFeature_RNA", "nCount_RNA")
p1_filtered=VlnPlot(input_sce.filt, group.by = "orig.ident", features = feats, pt.size = 0, ncol = 2) +
NoLegend()
w=length(unique(input_sce.filt$orig.ident))/3+5;w
ggsave(filename="Vlnplot1_filtered.pdf",plot=p1_filtered,width = w,height = 5)
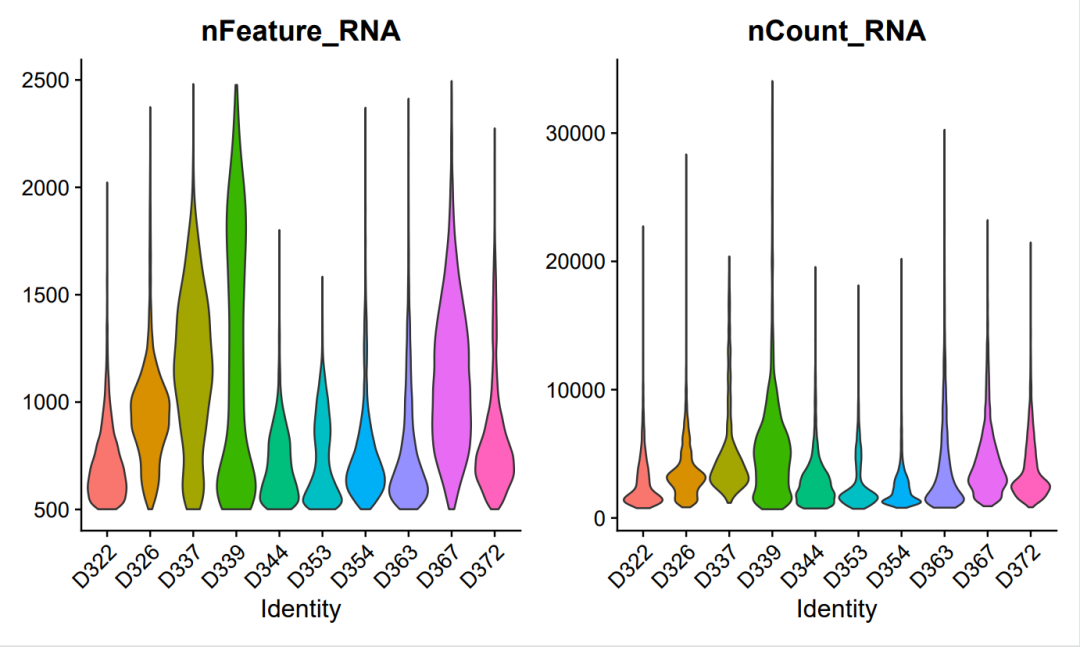
После фильтрации
Основное значение контроля качества: в каждом образце можно удалить некоторые гены со слишком высоким или слишком низким уровнем экспрессии.
Помимо визуализации nFeature_RNA и nCount_RNA в клетках во время основного процесса контроля качества, мы также будем визуализировать nFeature_RNA и nCount_RNA при выполнении кластеризации с уменьшением размерности.
Применение в кластеризации уменьшения размерности ячеек
При выборе соответствующего порогового значения для предварительного просмотра,мы будем использоватьcheck-all-markers.RСкрипт,Визуализируйте на основе общих генов-маркеров и рисуйте диаграммы umap.

существоватьcheck-all-markers.RСкрипт,Помогите нам проверить и подтвердить экспрессию генов в каждой субпопуляции клеток, тем самым помогая нам определить, является ли это двойной клеткой.。
Конкретные твиты:Как исключить двойные клетки
Когда мы просто называем подгруппы,Обычно выбирают более низкое разрешение 0,1.,ЧтосуществоватьGSE208706данныеиз0.1Группировкасередина,Мы ясно видим, что Группа 9 относительно длинная и узкая.,и содержит маркерные гены для двух разных субпопуляций клеток.。
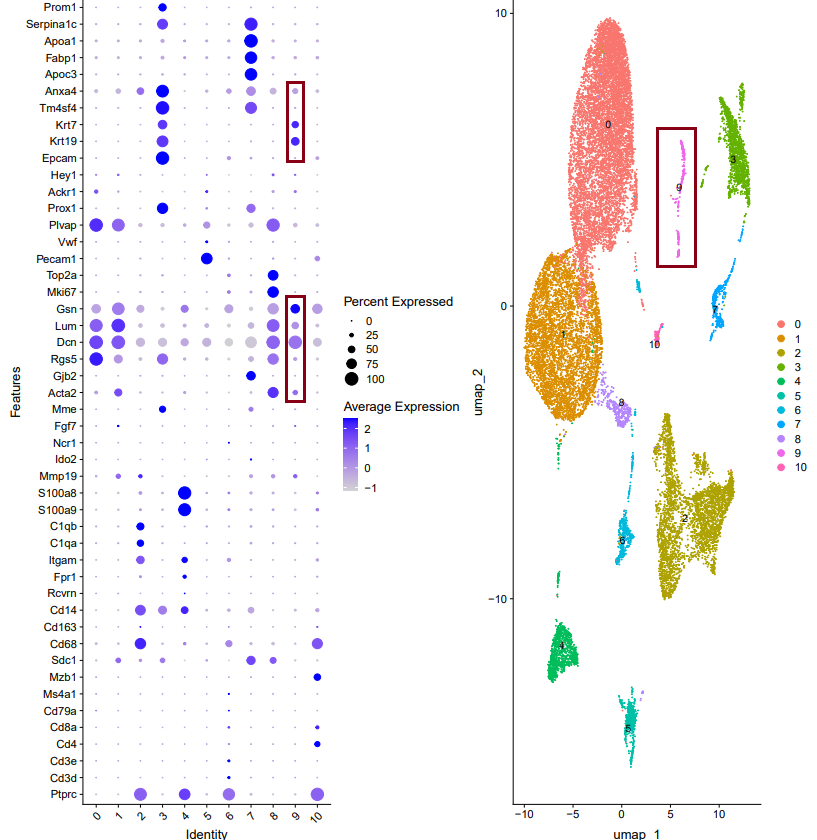
Чтобы определить, является ли это двойной ячейкой,Нам нужно объединитьОценивали общее количество РНК отдельных клеток в каждой субпопуляции.
if("percent_mito" %in% colnames(sce.all.int@meta.data ) ){
#Визуализация Указанное выше соотношение ячеек
feats <- c("nFeature_RNA", "nCount_RNA", "percent_mito", "percent_ribo", "percent_hb")
feats <- c("nFeature_RNA", "nCount_RNA")
p1=VlnPlot(sce.all.int , features = feats, pt.size = 0, ncol = 2) +
NoLegend()
w=length(unique(sce.all.int$orig.ident))/3+5;w
ggsave(filename=paste0(pro,"Vlnplot1.pdf"),plot=p1,width = w,height = 5)
feats <- c("percent_mito", "percent_ribo", "percent_hb")
p2=VlnPlot(sce.all.int, features = feats, pt.size = 0, ncol = 3, same.y.lims=T) +
scale_y_continuous(breaks=seq(0, 100, 5)) +
NoLegend()
w=length(unique(sce.all.int$orig.ident))/2+5;w
ggsave(filename=paste0(pro,"Vlnplot2.pdf"),plot=p2,width = w,height = 5)
}

Результат визуализации nFeature_RNA показал, что уровень экспрессии группы 8 был высоким, тогда как экспрессия группы 9 была нормальной.。на основеMarkerГенный вывод №.8Группа находится на стадии распространения.изклетка,Таким образом, высокий уровень экспрессии является разумным.
и после увеличения разрешения,ОбнаружитьГруппа 9 подразделяется на две подгруппы и не является двуклеточной.。
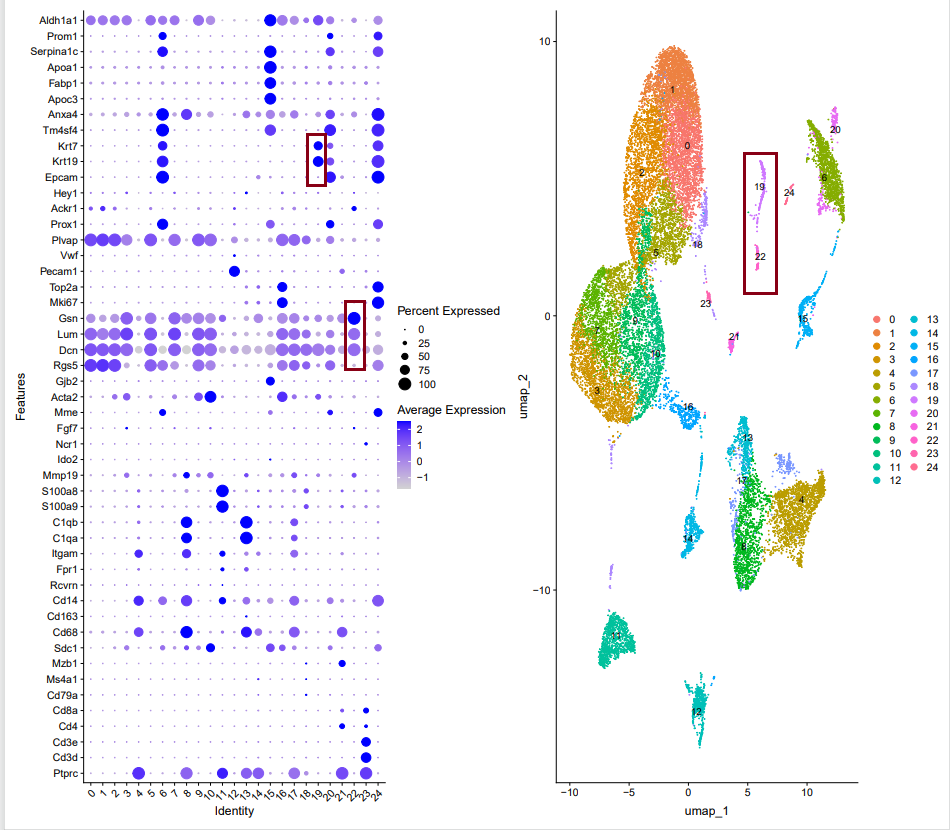
Обычно мыОн будет оцениваться на основе срединной линии и максимального значения, а затем разрешение будет увеличено, чтобы увидеть, разделены ли субпопуляции, а затем является ли это двойной ячейкой.
Соотношение митохондрий
На официальном сайте и в рамках нашего стандартного процесса контроля качества.,Можно рассчитатьСоотношение митохондрий
насизqc.RСкриптсередина Это верноСоотношение рибосом и эритроцитовБыли произведены расчетыи Визуализация,Давайте вместе узнаем об этом содержании в следующем выпуске!



Решение проблемы искажения китайских символов при чтении файлов Net Core.
Реализация легких независимых конвейеров с использованием Brighter

Как удалить и вернуть указанную пару ключ-значение из ассоциативного массива в PHP
Feiniu fnos использует Docker для развертывания учебного пособия по AList
Принципы и практика использования многопоточности в различных версиях .NET.
Как использовать PaddleOCRSharp в рамках .NET

CRUD используется уже два или три года. Как читать исходный код Spring?

Устраните проблему совместимости между версией Spring Boot и Gradle Java: возникла проблема при настройке корневого проекта «demo1» > Не удалось.

Научите вас шаг за шагом, как настроить Nginx.
Это руководство — все, что вам нужно для руководства по автономному развертыванию сервера для проектов Python уровня няни (рекомендуемый сборник).

Не удалось запустить docker.service — Подробное объяснение идеального решения ️

Настройка файлового сервера Samba в системе Linux Centos. Анализ NetBIOS (супер подробно)
Как настроить метод ssh в Git, как получить и отправить код через метод ssh
RasaGpt — платформа чат-ботов на основе Rasa и LLM.

Nomic Embed: воспроизводимая модель внедрения SOTA с открытым исходным кодом.

Улучшение YOLOv8: EMA основана на эффективном многомасштабном внимании, основанном на межпространственном обучении, и эффект лучше, чем у ECA, CBAM и CA. Малые цели имеют очевидные преимущества | ICASSP2023

Урок 1 серии Libtorch: Тензорная библиотека Silky C++

Руководство по локальному развертыванию Stable Diffusion: подробные шаги и анализ распространенных проблем
Полностью автоматический инструмент для работы с видео в один клик: VideoLingo

Улучшения оптимизации RT-DETR: облегченные улучшения магистрали | Support Paddle облегченный rtdetr-r18, rtdetr-r34, rtdetr-r50, rtdet
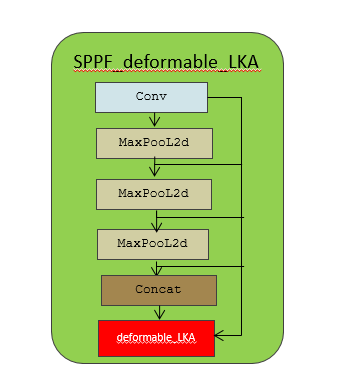
Эксклюзивное оригинальное улучшение YOLOv8: собственная разработка SPPF | Деформируемое внимание с большим ядром (D-LKA Attention), большое ядро свертки улучшает механизм внимания восприимчивых полей с различными функциями

Создано Datawhale: выпущено «Руководство по тонкой настройке развертывания большой модели GLM-4»!
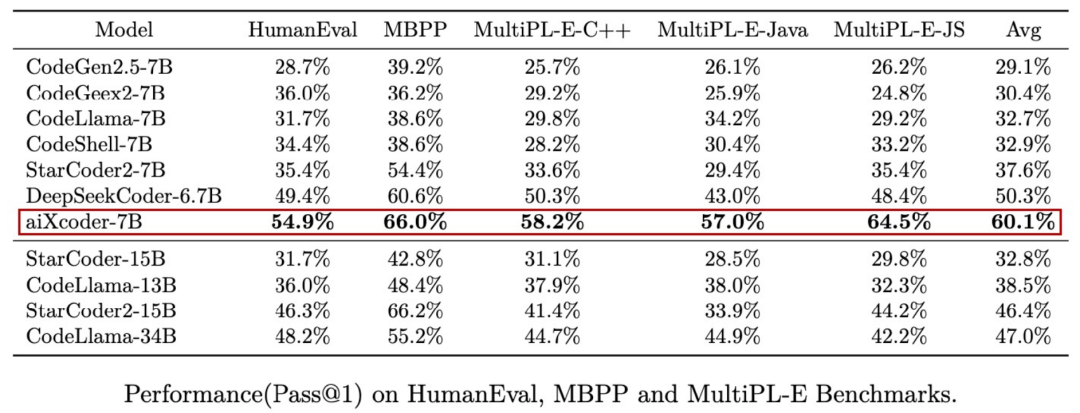
7B превышает десятки миллиардов, aiXcoder-7B с открытым исходным кодом Пекинского университета — это самая мощная модель большого кода, лучший выбор для корпоративного развертывания.
Используйте модель Huggingface, чтобы заменить интерфейс внедрения OpenAI в китайской среде.

Оригинальные улучшения YOLOv8: несколько новых улучшений | Сохранение исходной информации — алгоритм отделяемой по глубине свертки (MDSConv) |
Второй пилот облачной разработки | Быстро поиграйте со средствами разработки на базе искусственного интеллекта
Бесшовная интеграция, мгновенный интеллект [1]: платформа больших моделей Dify-LLM, интеграция с нулевым кодированием и встраивание в сторонние системы, более 42 тысяч звезд, чтобы стать свидетелями эксклюзивных интеллектуальных решений.

Решенная Ошибка | Загрузка PyTorch медленная: TimeoutError: [Errno 110] При загрузке факела истекло время ожидания — Cat Head Tiger

Brother OCR, библиотека с открытым исходным кодом для Python, которая распознает коды проверки.