CellTrek объединяет несколько методов сбора данных об отдельных ячейках и данных о простое
CellTrekОпубликовано в2022ежегодныйNature Biotechnology,под названием «Пространственное charting of single-cell transcriptomes in ткани». CellTrek может комбинировать одноклеточные и пространственные транскриптомные данные, чтобы точно определять расположение отдельных клеток в тканях и создавать пространственные атласы клеток. gitHub по адресу https://github.com/navinlabcode/CellTrek.
1. Рабочий процесс алгоритма CellTrek
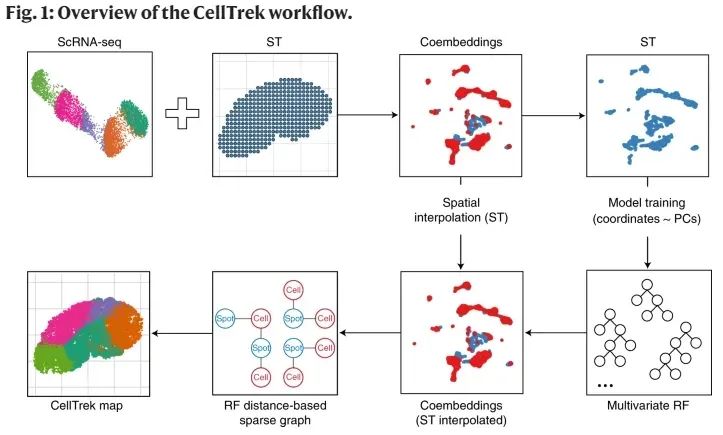
Рисунок 1: Обзор рабочего процесса CellTrek.
- CellTrek сначала объединяет исходные матрицы ST и scRNA-seq.,Затем объединенная матрица уменьшается по размеру. Затем извлеките данные пространственного транскриптома, чтобы построить многомерную модель случайного леса (RF).,где пространственные координаты являются результатом,Скрытые особенности являются предикторами;
- Выполните двумерную пространственную интерполяцию на STданных, чтобы улучшить пятна ST. Затем,Применить обученную RFModel к встроенным данным (интерполяция ST),для создания матрицы расстояний RF,Эта матрица будетиспользоватьпара ближайших соседей(MNN)Преобразовать в разреженный граф;
- наконецна основеразреженный граф,Вложение данных одноклеточного транскриптома в RF-модель,Постройте матрицу сходства выражений Spot-Cell,Добавьте информацию о пространственных координатах к каждому результату отдельной ячейки и включите предварительное отображение.
как мы все знаем,Одним из недостатков 10-кратного увеличения данных в режиме ожидания не является разрешение одной ячейки.,Скорее, это точка смешения, содержащая более десяти ячеек.。Алгоритм CellTrek может в полной мере использовать данные транскриптома одной клетки и сопоставлять информацию одной клетки с пространственными срезами транскриптома. . Примечательно, что CellTrek может обеспечить разрешение простоя данных в одной ячейке, в отличие от предыдущих методов деконволюции.
2. Реализация кода CellTrek
Установка CellTrek относительно проста:
library(devtools)
install_github("navinlabcode/CellTrek")
Загрузка пакета R:
library(Seurat)
library(SeuratData)
library(ggplot2)
library(patchwork)
library(dplyr)
library(readr)
library(CellTrek)
library(viridis)
library(ConsensusClusterPlus)
library(SeuratObject)
Шаг 1. Загрузите образцы данных.
brain_st_cortex <- read_rds("./Rawdata/brain_st_cortex.rds")
brain_sc <- read_rds("./Rawdata/brain_sc.rds")
## Rename the cells/spots with syntactically valid names
brain_st_cortex <- RenameCells(brain_st_cortex, new.names=make.names(Cells(brain_st_cortex)))
brain_sc <- RenameCells(brain_sc, new.names=make.names(Cells(brain_sc)))
Визуализация данных холостого хода:
## Visualize the ST data
SpatialDimPlot(brain_st_cortex)
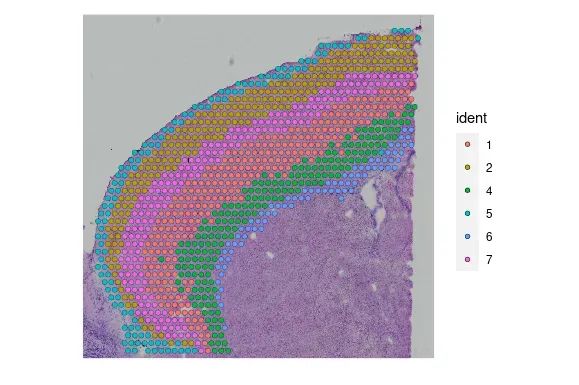
image-20231105144510038
Визуализируйте данные одной ячейки:
## Visualize the scRNA-seq data
DimPlot(brain_sc, label = T, label.size = 4.5)
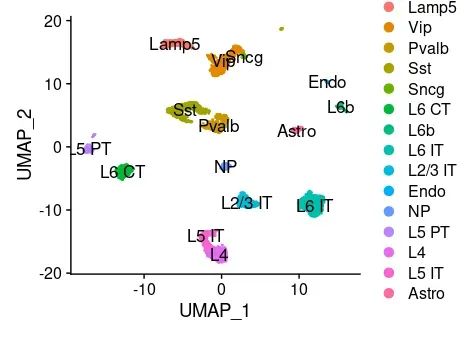
image-20231105144526583
Шаг 2. Используйте CellTrek для сопоставления ячеек.
#### 2. использовать CellTrek Выполнить сопоставление ячеек
# We first co-embed ST and scRNA-seq datasets using traint
brain_traint <- CellTrek::traint(st_data=brain_st_cortex,
sc_data=brain_sc,
sc_assay='RNA',
cell_names='cell_type')
## Мы можем проверить результаты совместного внедрения, чтобы увидеть, есть ли совпадение между двумя модальностями данных.
DimPlot(brain_traint, group.by = "type")
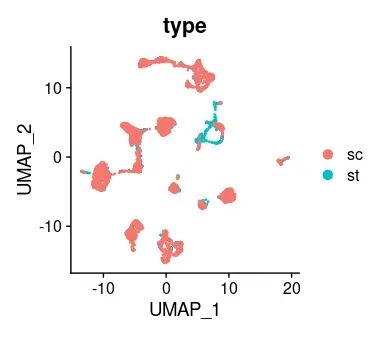
image-20231105144622926
# После совместного внедрения мы можем сопоставить отдельные ячейки с их пространственным расположением.
# здесь,Мы используем нелинейную интерполяцию (intp = T, intp_lin=F) метод усиления пятен ST
brain_celltrek <- CellTrek::celltrek(st_sc_int=brain_traint, int_assay='traint', sc_data=brain_sc, sc_assay = 'RNA',
reduction='pca', intp=T, intp_pnt=5000, intp_lin=F, nPCs=30, ntree=1000,
dist_thresh=0.55, top_spot=5, spot_n=5, repel_r=20, repel_iter=20, keep_model=T)$celltrek
# После завершения сопоставления ячеек,нас可以использовать celltrek_vis интерактивный Визуализация CellTrek результат
brain_celltrek$cell_type <- factor(brain_celltrek$cell_type,
levels=sort(unique(brain_celltrek$cell_type)))
CellTrek::celltrek_vis(brain_celltrek@meta.data %>%
dplyr::select(coord_x, coord_y, cell_type:id_new),
brain_celltrek@images$anterior1@image,
brain_celltrek@images$anterior1@scale.factors$lowres)
Шаг 3. Анализ совместной локализации клеток CellTrek.
#### 3. Анализ клеточной колокализации
# на основе CellTrek результат,нас可以использовать SColoc Обобщить закономерности колокализации между различными типами клеток.
# здесь,В качестве примера мы используем тип клеток глутаматергических нейронов (рекомендуется удалить некоторые типы клеток).,Например,n<20,Типы клеток с очень небольшим количеством клеток)。
# Сначала мы выделили типы глутаматергических нейрональных клеток на основе результатов нашего картирования.
glut_cell <- c('L2/3 IT', 'L4', 'L5 IT', 'L5 PT', 'NP', 'L6 IT', 'L6 CT', 'L6b')
names(glut_cell) <- make.names(glut_cell)
brain_celltrek_glut <- subset(brain_celltrek, subset=cell_type %in% glut_cell)
#Thenusescoloc для анализа совместной локализации:
brain_celltrek_glut$cell_type <- factor(brain_celltrek_glut$cell_type,
levels=glut_cell)
## нас从图中提取最小生成树(MST)результат。
brain_sgraph_KL_mst_cons <- brain_sgraph_KL$mst_cons
rownames(brain_sgraph_KL_mst_cons) <- colnames(brain_sgraph_KL_mst_cons) <- glut_cell[colnames(brain_sgraph_KL_mst_cons)]
## Затем мы извлекаем метаданные (включая типы ячеек и информацию об их частоте).
brain_cell_class <- brain_celltrek@meta.data %>% dplyr::select(id=cell_type) %>% unique
brain_celltrek_count <- data.frame(freq = table(brain_celltrek$cell_type))
brain_cell_class_new <- merge(brain_cell_class, brain_celltrek_count, by.x ="id", by.y = "freq.Var1")
brain_sgraph_KL <- CellTrek::scoloc(brain_celltrek_glut,
col_cell='cell_type',
use_method='KL', eps=1e-50)
CellTrek::scoloc_vis(brain_sgraph_KL_mst_cons,
meta_data=brain_cell_class_new )
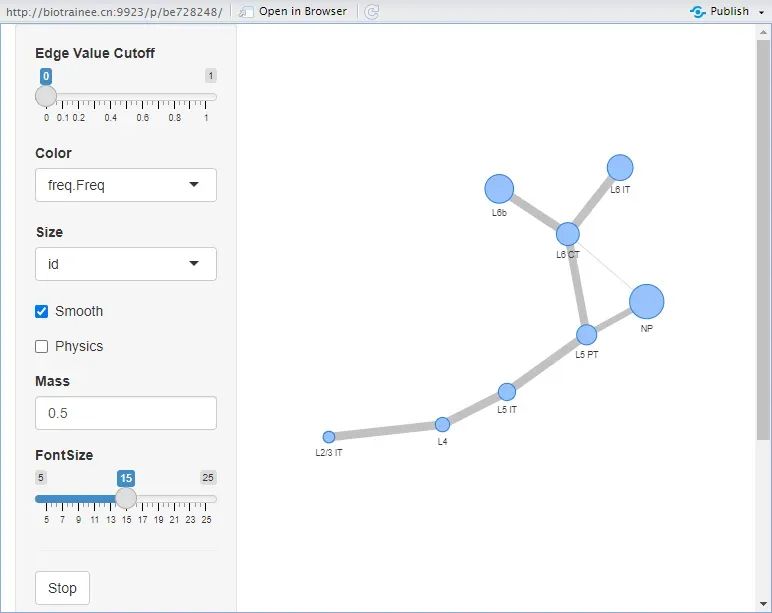
image-20231105145004137
Шаг 4. Пространственно-взвешенный анализ совместной экспрессии типов клеток-мишеней.
# на основеCellTrekрезультат,нас可以использоватьSCoexpМодули для дальнейшего изучения закономерностей совместной экспрессии в интересующих типах клеток。
# здесь,нас将использоватьконсенсусная кластеризация(CC)метод сL5 Возьмем, к примеру, ИТ-подразделение. Сначала извлеките L5 из результатов диаграммы. ИТ-ячейка.
brain_celltrek_l5 <- subset(brain_celltrek, subset=cell_type=='L5 IT')
brain_celltrek_l5@assays$RNA@scale.data <- matrix(NA, 1, 1)
brain_celltrek_l5$cluster <- gsub('L5 IT VISp ', '', brain_celltrek_l5$cluster)
DimPlot(brain_celltrek_l5, group.by = 'cluster')
# Мы выбираем 2000 лучших вариабельных генов (исключая митохондрии, рибосомы и гены с высоким нулевым значением).
brain_celltrek_l5 <- FindVariableFeatures(brain_celltrek_l5)
vst_df <- brain_celltrek_l5@assays$RNA@meta.features %>% data.frame %>% mutate(id=rownames(.))
nz_test <- apply(as.matrix(brain_celltrek_l5[['RNA']]@data), 1, function(x) mean(x!=0)*100)
hz_gene <- names(nz_test)[nz_test<20]
mt_gene <- grep('^Mt-', rownames(brain_celltrek_l5), value=T)
rp_gene <- grep('^Rpl|^Rps', rownames(brain_celltrek_l5), value=T)
vst_df <- vst_df %>% dplyr::filter(!(id %in% c(mt_gene, rp_gene, hz_gene))) %>% arrange(., -vst.variance.standardized)
feature_temp <- vst_df$id[1:2000]
# насиспользовать scoexp Был проведен пространственно-взвешенный анализ совместной экспрессии генов.
brain_celltrek_l5_scoexp_res_cc <- CellTrek::scoexp(celltrek_inp=brain_celltrek_l5,
assay='RNA',
approach='cc',
gene_select = feature_temp,
sigm=140,
avg_cor_min=.4,
zero_cutoff=3,
min_gen=40, max_gen=400)
# нас可以использоватьтепловая карта Визуализациямодуль коэкспрессии。
brain_celltrek_l5_k <- rbind(data.frame(gene=c(brain_celltrek_l5_scoexp_res_cc$gs[[1]]), G='K1'),
data.frame(gene=c(brain_celltrek_l5_scoexp_res_cc$gs[[2]]), G='K2')) %>%
magrittr::set_rownames(.$gene) %>% dplyr::select(-1)
pheatmap::pheatmap(brain_celltrek_l5_scoexp_res_cc$wcor[rownames(brain_celltrek_l5_k), rownames(brain_celltrek_l5_k)],
clustering_method='ward.D2', annotation_row=brain_celltrek_l5_k, show_rownames=F, show_colnames=F,
treeheight_row=10, treeheight_col=10, annotation_legend = T, fontsize=8,
color=viridis(10), main='L5 IT spatial co-expression')
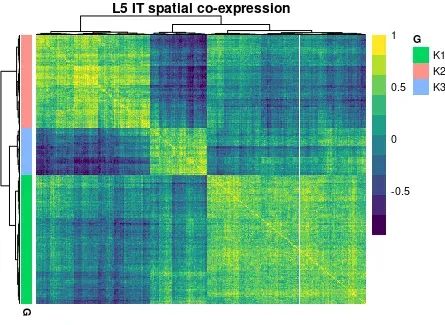
image-20231105151101908
# нас确定了3разные генные модули。на основенас识别的共表达模块,Можно подсчитать баллы по модулям.
brain_celltrek_l5 <- AddModuleScore(brain_celltrek_l5,
features=brain_celltrek_l5_scoexp_res_cc$gs,
name='CC_', nbin=10, ctrl=50, seed=42)
## Визуализация1
FeaturePlot(brain_celltrek_l5,
grep('CC_', colnames(brain_celltrek_l5@meta.data),
value=T), ncol = 1)
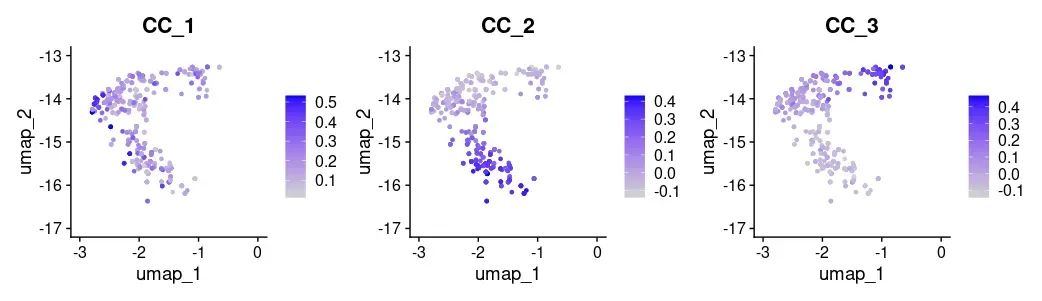
image-20231105151156465
## Визуализация2
SpatialFeaturePlot(brain_celltrek_l5,
grep('CC_', colnames(brain_celltrek_l5@meta.data), value=T))
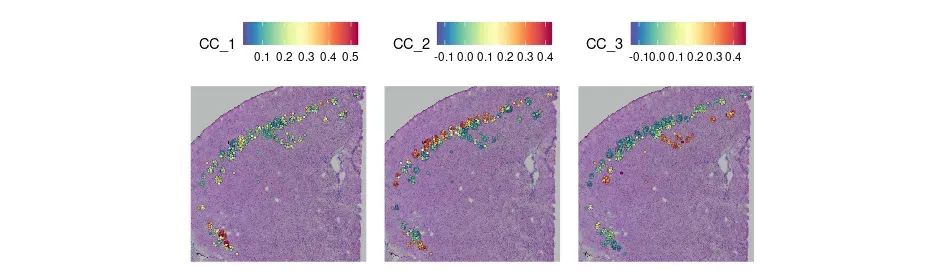
image-20231105151243508
3. Пасхальные яйца
Ввиду критики со стороны некоторых читателей о том, что мы лишь «копируем английское Учебное» пособие”,«Копируйте английские посты»,здесь яна основеверноCellTrekПонимание воспроизведено на рисунке ниже.E(Этого нет в официальном руководстве).
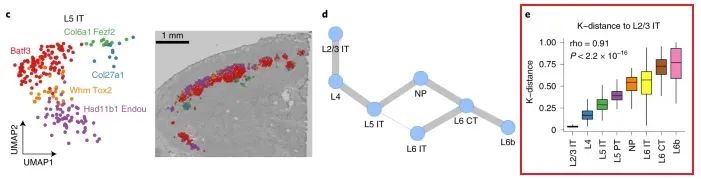
image-20231105151822596
На рисунке E рассчитывается пространственное расстояние между целевым типом клеток (L2/3T) и другими типами клеток. Результаты показывают, что субпопуляция клеток L4 пространственно ближе всего к L2/3T (см. рисунки C-E, рисунок D воспроизведен выше):
Первым шагом является получение информации о типе ячейки и пространственных координатах:
# coord_x и coord_y — это пространственные координаты:
inp_df <- brain_celltrek_glut@meta.data %>% dplyr::select(cell_names = dplyr::one_of('cell_type'),
coord_x, coord_y)
inp_df$coord_x = 270-inp_df$coord_x
head(inp_df)
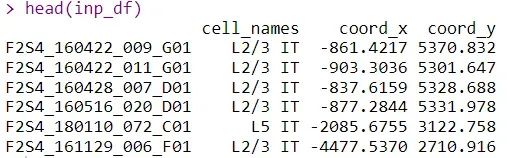
image-20231105164435672
Заинтересованные друзья смогут понять, зачем нужен «270-inp_df$coord_x».
Второй шаг — вычислить кластеризацию целевой ячейки и других ячеек:
output <- kdist(inp_df = inp_df,
ref = "L2/3 IT", #тип целевой ячейки
ref_type = 'all',
que = glut_cell, #остальные клетки
k = 10,
new_name = "L23ITvs.Others",
keep_nn = F)
head(output$kdist_df) #наконецрезультат
# L23ITvs.Others
# F2S4_160422_009_G01 59.72424
# F2S4_160422_011_G01 62.78655
# F2S4_160428_007_D01 58.06266
# F2S4_160516_020_D01 53.70867
# F2S4_180110_072_C01 376.52573
# F2S4_161129_006_F01 757.52189
res = output$kdist_df
res$barcode = row.names(res)
inp_df$barcode = row.names(inp_df)
res = left_join(res, inp_df)
head(res)
# L23ITvs.Others barcode cell_names coord_x coord_y
# 1 59.72424 F2S4_160422_009_G01 L2/3 IT -861.4217 5370.832
# 2 62.78655 F2S4_160422_011_G01 L2/3 IT -903.3036 5301.647
# 3 58.06266 F2S4_160428_007_D01 L2/3 IT -837.6159 5328.688
# 4 53.70867 F2S4_160516_020_D01 L2/3 IT -877.2844 5331.978
# 5 376.52573 F2S4_180110_072_C01 L5 IT -2085.6755 3122.758
# 6 757.52189 F2S4_161129_006_F01 L2/3 IT -4477.5370 2710.916
Третий шаг, визуализация:
library(ggpubr)
ggboxplot(data = res,
x = "cell_names",
y = "L23ITvs.Others",
fill = "cell_names",
title = "K-distance to L2/3 IT cells")+
stat_compare_means(method = "kruskal.test") +
theme(plot.title = element_text(color="black",hjust = 0.5),
axis.text.x = element_text(angle = 90, hjust = 1,vjust = 0.5), #,vjust = 0.5
legend.position = "none") + labs(y = "Distance")
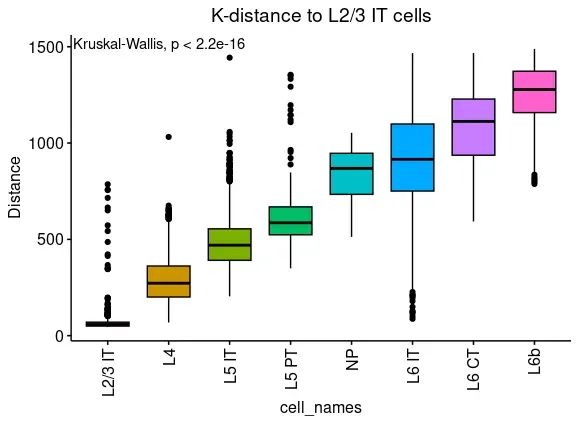
image-20231105165306137
В принципе, то же самое, что и исходный текст.
Позвольте мне объяснить немного больше:
- На самом деле «Английское Учебное пособие”,Само по себе имеет некоторые трудности для небольшого количества друзей.,Трудности при чтении второго,В основном речь идет о понимании кода;
- Вторая небольшая проблема заключается в том, что некоторое «Учебное пособие по английскому языку» В "пособии" много ошибок. Наши друзья в дереве навыков обязательно проверят его еще раз при написании постов. код пособия, если есть ошибки, мы поможем их решить, а затем запишем решение в Учебное пособиевнутри,Вы должны знать, что некоторые ошибки не могут быть устранены некоторыми друзьями в течение нескольких дней или даже недель, поэтому само по себе «копирование уроков английского языка» не будет простым делом.;
- Третий,Большая часть Учебного пособия,Мы все добавим нашу собственную систему понимания и знаний для организации и объединения,Это нормально, когда что-то не так,Мы очень приветствуем дружеские дискуссии и вопросы.,Я надеюсь улучшить себя, задавая вопросы другим,Это также одно из моих первоначальных намерений поделиться своими заметками;
- Наконец, написание постов действительно не приносит денег... В основном мы зарабатываем на любви (честно говоря, написание постов у меня обычно занимает много времени, иногда даже день-два. За это время я могу заработать много денег...). Поэтому, пожалуйста, дорожите нашими друзьями по дереву навыков...
- END -


Учебное пособие по Jetpack Compose для начинающих, базовые элементы управления и макет

Код js веб-страницы, фон частицы, код спецэффектов

【новый! Суперподробное】Полное руководство по свойствам компонентов Figma.
🎉Обязательно к прочтению новичкам: полное руководство по написанию мини-программ WeChat с использованием программного обеспечения Cursor.
[Забавный проект Docker] VoceChat — еще одно приложение для мгновенного чата (IM)! Может быть встроен в любую веб-страницу!

Как реализовать переход по странице в HTML (html переходит на указанную страницу)

Как решить проблему зависания и низкой скорости при установке зависимостей с помощью npm. Существуют ли доступные источники npm, которые могут решить эту проблему?

Серия From Zero to Fun: Uni-App WeChat Payment Practice WeChat авторизует вход в систему и украшает страницу заказа, создает интерфейс заказа и инициирует запрос заказа

Серия uni-app: uni.navigateЧтобы передать скачок значения

Апплет WeChat настраивает верхнюю панель навигации и адаптируется к различным моделям.

JS-время конвертации

Обеспечьте бесперебойную работу ChromeDriver 125: советы по решению проблемы chromedriver.exe не найдены

Поле комментария, щелчок мышью, специальные эффекты, js-код
Объект массива перемещения объекта JS

Как открыть разрешение на позиционирование апплета WeChat_Как использовать WeChat для определения местонахождения друзей
Я даю вам два набора из 18 простых в использовании фонов холста Power BI, так что вам больше не придется возиться с цветами!

Получить текущее время в js_Как динамически отображать дату и время в js

Вам необходимо изучить сочетания клавиш vsCode для форматирования и организации кода, чтобы вам больше не приходилось настраивать формат вручную.

У ChatGPT большое обновление. Всего за 45 минут пресс-конференция показывает, что OpenAI сделал еще один шаг вперед.
Copilot облачной разработки — упрощение разработки

Микросборка xChatGPT с низким кодом, создание апплета чат-бота с искусственным интеллектом за пять шагов

CUDA Out of Memory: идеальное решение проблемы нехватки памяти CUDA
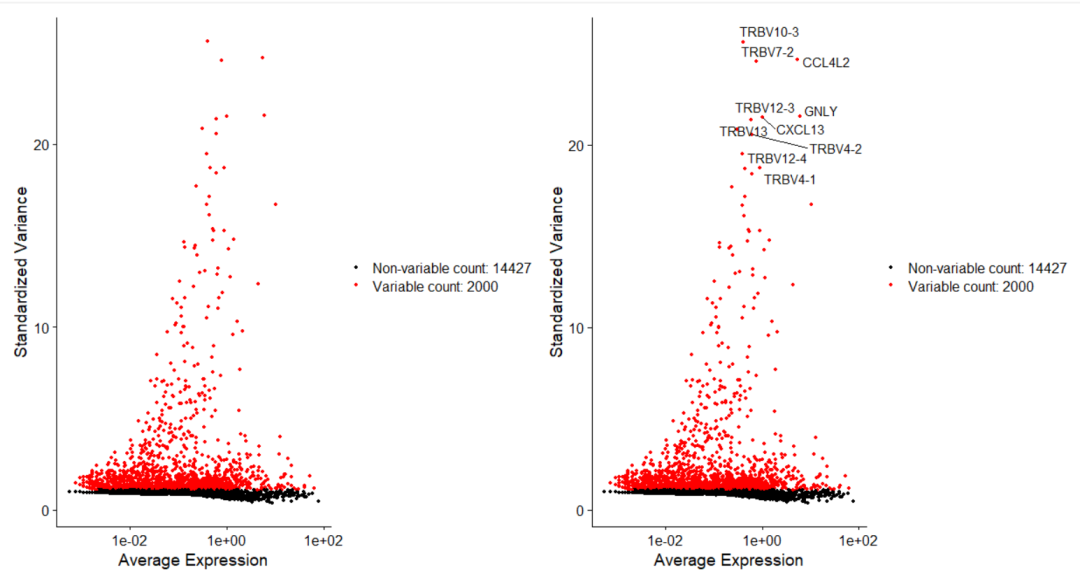
Анализ кластеризации отдельных ячеек, который должен освоить каждый&MarkerгенетическийВизуализация

vLLM: мощный инструмент для ускорения вывода ИИ

CodeGeeX: мощный инструмент генерации кода искусственного интеллекта, который можно использовать бесплатно в дополнение к второму пилоту.

Машинное обучение Реальный бой LightGBM + настройка параметров случайного поиска: точность 96,67%
Бесшовная интеграция, мгновенный интеллект [1]: платформа больших моделей Dify-LLM, интеграция без кодирования и встраивание в сторонние системы, более 42 тысяч звезд, чтобы стать свидетелями эксклюзивных интеллектуальных решений.
LM Studio для создания локальных больших моделей
Как определить количество слоев и нейронов скрытых слоев нейронной сети?
