Анализ кластеризации отдельных ячеек, который должен освоить каждый&MarkerгенетическийВизуализация
В течение года последние версии SeuratObject и Seurat V5 никому не понадобятся, так что вернитесь к v4...
Пакетное секвенирование транскриптома может предоставить информацию об общей экспрессии генов в ткани или клеточной линии, но не может решить проблему гетерогенности между различными клетками. Секвенирование отдельных клеток может предоставить характеристики экспрессии генов в каждой отдельной клетке, что позволяет исследователям изучать типы клеток. Более подробно; молекулярные характеристики, такие как статус клеток и клеточные субпопуляции. Поскольку данные секвенирования отдельных клеток очень неоднородны, для обработки и интерпретации данных требуются специальные методы анализа данных, такие как кластерный анализ, алгоритмы уменьшения размерности и т. Д. Это означает, что анализ секвенирования отдельных клеток более сложен, чем анализ массового транскриптома.
Прежде чем стать учеником в дереве навыков «Создание»,Я никогда не подвергался анализу секвенирования отдельных клеток.,мне повезло сегодняи Учитесь у прекрасных учителей,Я также очень рад возможности распространить полученные знания среди большего количества новичков.,Давайте вместе станем свидетелями роста друг друга! ХОРОШО,Ближе к дому,Сегодня мы впервые узнаем об анализе кластеризации отдельных ячеек с г-ном Цзяньмином.&Markerгенетический Визуализация。
Если взять в качестве примера общедоступный набор данных GSE182434, код и результаты будут следующими:
#Установить программное обеспечение
rm(list=ls())
options(stringsAsFactors = F)
getwd()
library(Seurat)
library(ggplot2)
#install.packages(c('clustree'))
library(clustree)
library(cowplot)
library(dplyr)
library(patchwork)
#Читать данные матикса
library(data.table)
dat=fread( "GSE182434_raw_count_matrix.txt.gz",data.table = F)
dim(dat)
dat[1:4,1:4]
rownames(dat)=dat[,1]
dat=dat[,-1]
dat[1:4,1:4]
annotation = fread( "GSE182434_cell_annotation.txt.gz",data.table = F)
annotation[1:4,1:4]
table(annotation$Patient,annotation$CellType)
#Образцы пациентов, связанные с скринингом (DLBCL002, DLBCL007, DLBCL008, DLBCL111)
#Условия фильтра ①、Четыре образца: DLBCL002、DLBCL007、 DLBCL008、DLBCL111
#Условия фильтрации ②, клетки CD8+
sample = c("DLBCL002","DLBCL007","DLBCL008","DLBCL111")
CellType = "T cells CD8"
annotation = annotation[annotation$Patient %in% sample,]
annotation = annotation[annotation$CellType %in% CellType,]
dim(annotation) Образцы № 4, всего 5000 клеток CD8.
dat = dat[,annotation$ID]
dat[1:5,1:5]
dim(dat)
View(dat)
View(annotation)
#Создаем объект Сёра
library(Seurat)
sce <- CreateSeuratObject(counts = dat,
project = "sce", #Название проекта
min.cells = 3,
min.features = 200)
sce
View(sce$nCount_RNA)
View(sce$nFeature_RNA)
View(sce$orig.ident)
sce$orig.ident<-annotation$Patient #Изменить сцену Информация о группировке образцов матрицы
View(sce$orig.ident)
#Определить гены с сильно варьирующими уровнями экспрессии
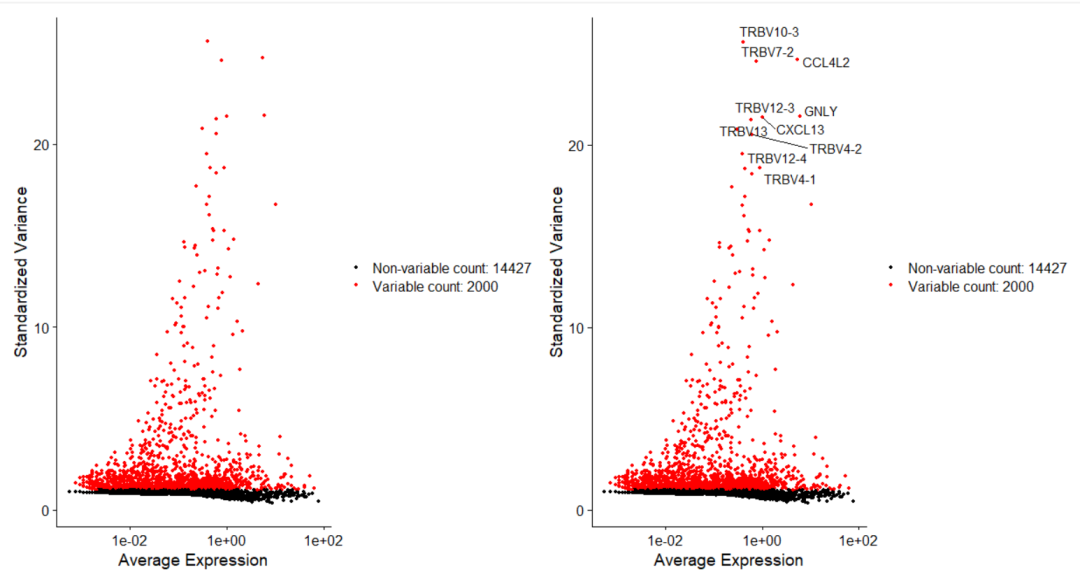
sce = FindVariableFeatures(sce,selection.method = "vst", nfeatures = 2000)#Они высоко экспрессируются в одних клетках и низки в других, что при дальнейшем анализе фокусируется на Эти гены помогают выделить биологические сигналы, сконцентрированные в отдельных клетках.
#существовать Seurat Процедура в , улучшена предыдущая версия за счет прямого моделирования отношений среднего значения и дисперсии, присущих данным одной ячейки, и реализована в функции FindVariableFeatures(). По умолчанию возвращается для каждого набора данных. 2,000 Характеристические гены использовали для последующего анализа.
Определите 10 основных гипервариабельных генов.
top10 <- head(VariableFeatures(sce), 10)
Нарисуйте карту гипервариабельных генов без надписей.
plot1 <- VariableFeaturePlot(sce)
Добавить десять лучших тегов
plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE)
plot1 + plot2
#Просто нарисуйте DimPlot, чтобы определить, требуется ли гармония
sce = NormalizeData(sce, normalization.method = "LogNormalize", scale.factor = 10000)
sce = FindVariableFeatures(sce)
sce = ScaleData(sce, vars.to.regress = c("nFeature_RNA", "percent_mito"))
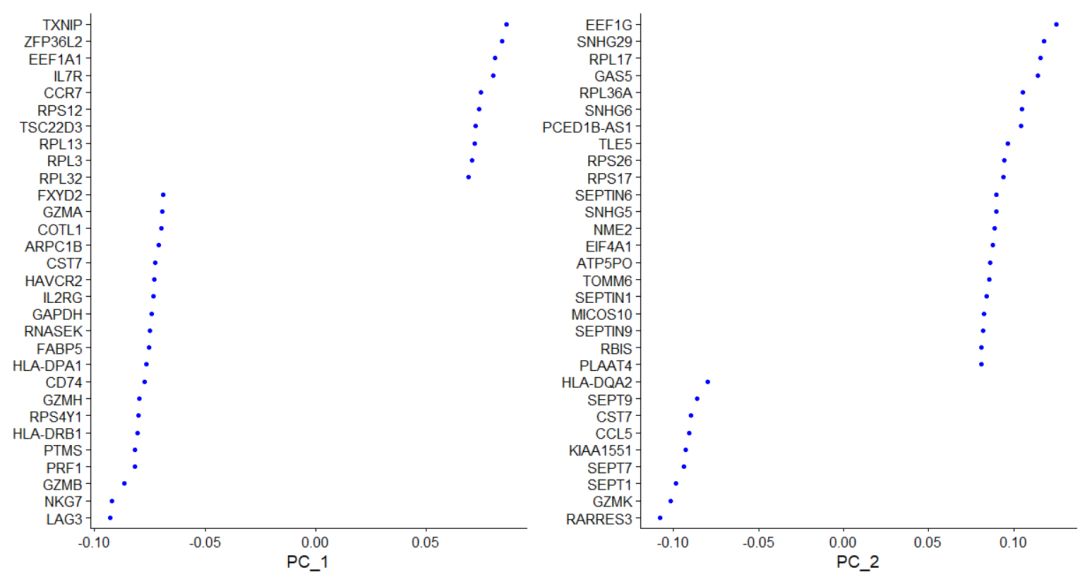
sce = RunPCA(sce, npcs = 20) #PCA уменьшение размерности, уменьшение размерности сильно вариабельных генов.
print(sce [["pca"]], dims = 1:5, nfeatures = 5)#Показать часть результатов
VizDimLoadings(sce , dims = 1:2, reduction = "pca")##Отображается в формате точечного изображения.
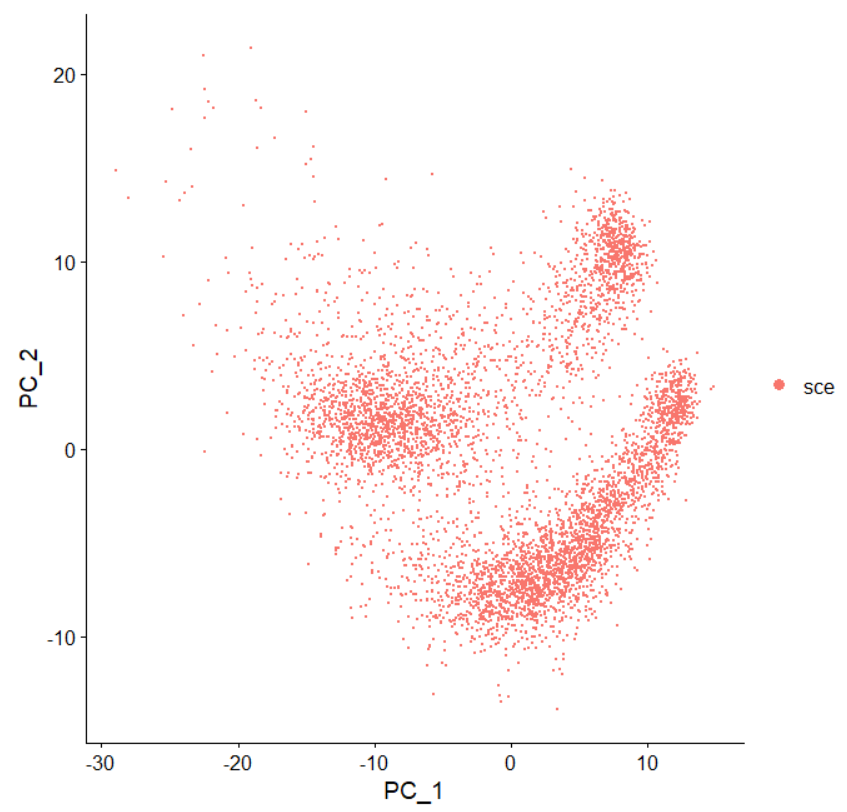
DimPlot(sce, reduction = "pca")#Проецируемая карта уменьшения размерности
DimHeatmap(sce, dims = 1:15, cells = 500, balanced = ВЕРНО) #DimHeatmap() позволяет легко исследовать основные источники неоднородности в концентрации данных и при попытке решить, какие из них включить. PC Очень полезно для дальнейшего последующего анализа. Ячейки и функции основаны на их PCA Сортировка баллов.

##Нелинейное уменьшение размерности
#Seurat Предоставляются различные методы нелинейного уменьшения размерности, например. tSNE и UMAP,Изучите эти эпизоды данных с помощью предварительной визуализации. Целью этих алгоритмов является изучение основного множества данных.,для того, чтобы разместить подобные ячейки в низкомерном пространстве. Ячейки в кластерах на основе графов, указанных выше, должны быть расположены рядом на этих графиках уменьшения размерности. как UMAP и tSNE вход, мы рекомендуем использовать тот же PC в качестве входных данных для кластерного анализа.
sce = RunTSNE(sce, npcs = 20)
sce = Runtsne(sce, dims = 1:10)
sce = RunUMAP(sce, dims = 1:10)
sce = FindNeighbors(sce, dims = 1:20, k.param = 60, prune.SNN = 1/15) #Этот димс должен соответствовать NPC PCA.
DimPlot(sce,reduction = "umap",label=T,group.by = "orig.ident")
ggsave(filename="no_harmony_DimPlot.pdf")
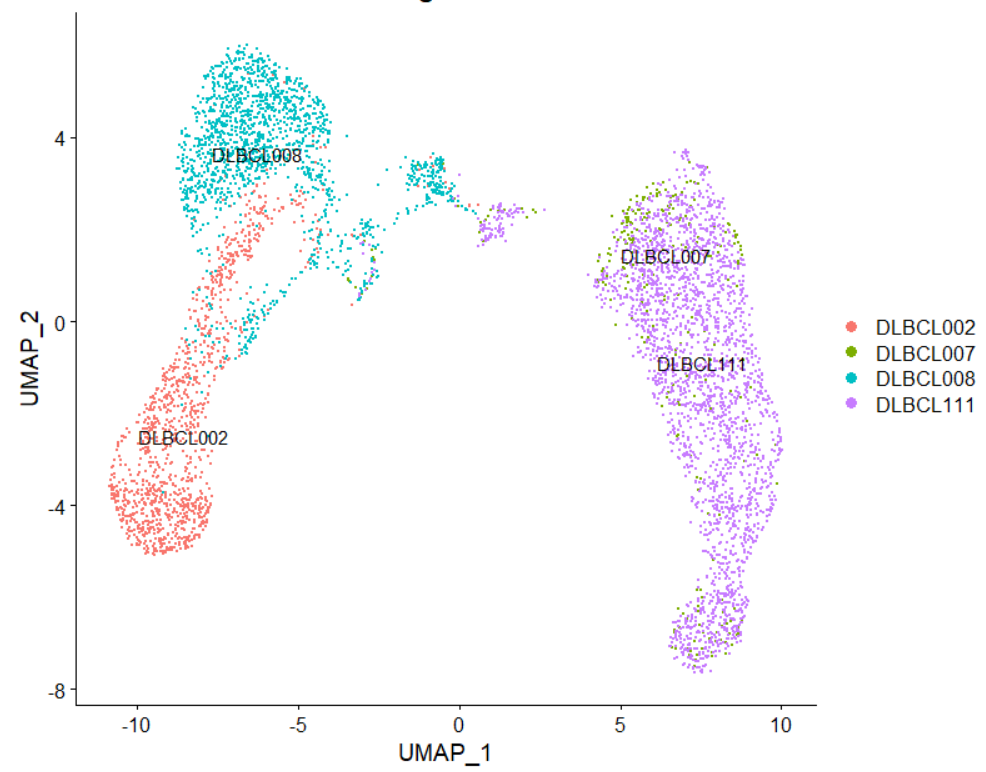
#Чтобы попасть в партию, нужна гармония, потому что активно проверялось 4 образца.
library(harmony)
sce <- RunHarmony(sce, "orig.ident")
names(sce@reductions)
sce = RunUMAP(sce, dims = 1:15, reduction = "harmony")
sce = RunTSNE(sce, npcs = 20,reduction = "harmony")
sce = FindNeighbors(sce, reduction = "harmony", dims = 1:15)
DimPlot(sce,reduction = "umap",label=T,group.by = "orig.ident")
ggsave(filename="harmony_DimPlot.pdf")
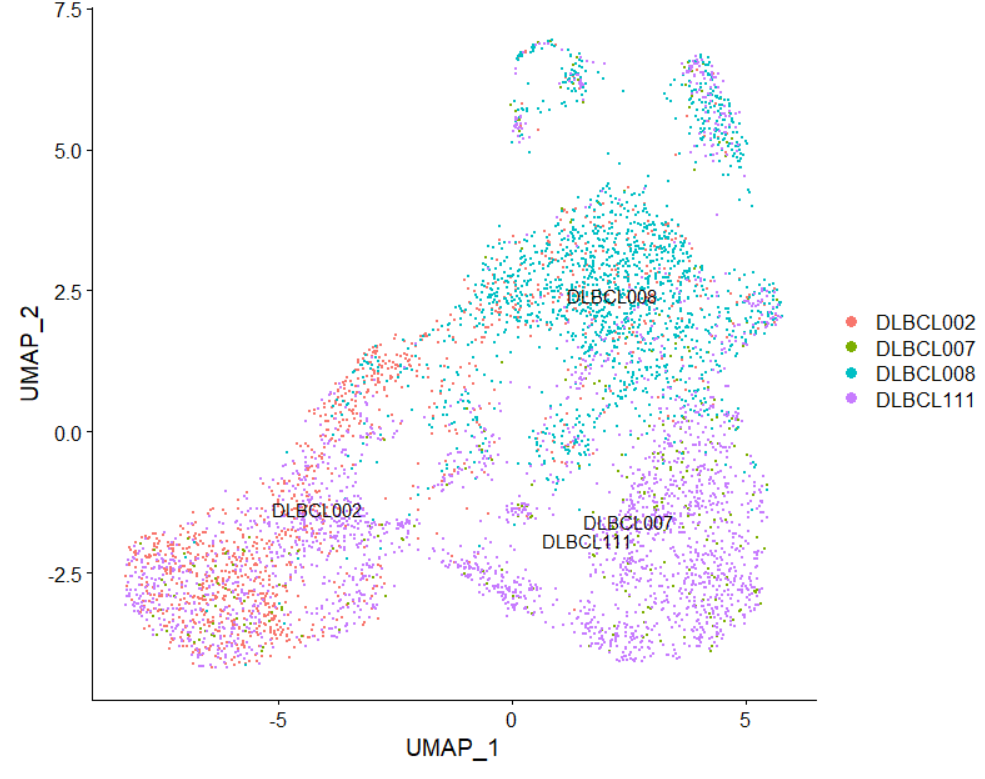
##(1)Установить разрешение
#Классификация разрешения
#Статья разделена на 6 групп
#Установите разные разрешения и наблюдайте за эффектом группировки (какое выбрать?)
for (res in c(0.01, 0.05, 0.1,0.15, 0.2, 0.3, 0.4,0.45,0.5,0.8,0.9,1)) {
sce=FindClusters(sce, resolution = res, algorithm = 1)
}#Классификация ячеек
colnames(sce@meta.data)
apply(sce@meta.data[,grep("RNA_snn_res",colnames(sce@meta.data))],2,table)
#0.8
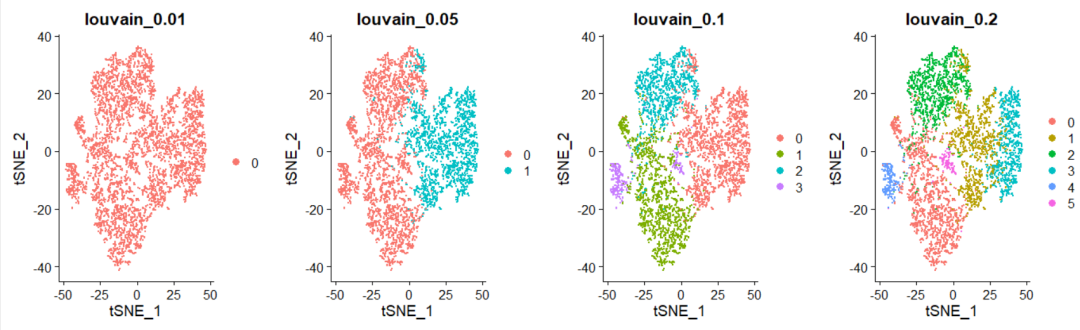
(2) Визуализируйте ситуацию группировки высокого и низкого разрешения.
p1_dim = plot_grid(ncol = 4, DimPlot(sce, reduction = "tsne", group.by = "RNA_snn_res.0.01") +
ggtitle("louvain_0.01"), DimPlot(sce, reduction = "tsne", group.by = "RNA_snn_res.0.05") +
ggtitle("louvain_0.05"), DimPlot(sce, reduction = "tsne", group.by = "RNA_snn_res.0.1") +
ggtitle("louvain_0.1"), DimPlot(sce, reduction = "tsne", group.by = "RNA_snn_res.0.2") +
ggtitle("louvain_0.2"))
p1_dim
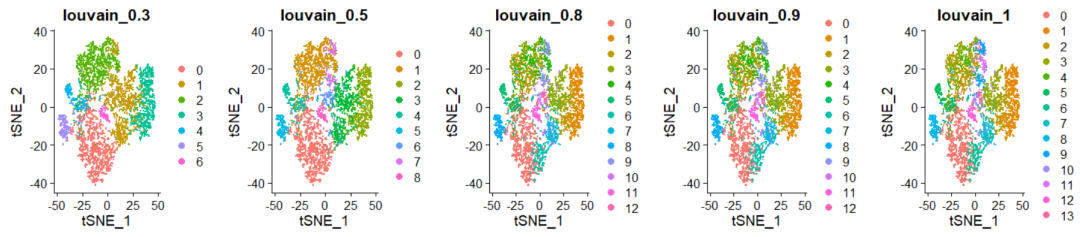
p1_dim = plot_grid(ncol = 5, DimPlot(sce, reduction = "tsne", group.by = "RNA_snn_res.0.3") +
ggtitle("louvain_0.3"),DimPlot(sce, reduction = "tsne", group.by = "RNA_snn_res.0.5") +
ggtitle("louvain_0.5"),DimPlot(sce, reduction = "tsne", group.by = "RNA_snn_res.0.8") +
ggtitle("louvain_0.8"), DimPlot(sce, reduction = "tsne", group.by = "RNA_snn_res.0.9") +
ggtitle("louvain_0.9"),DimPlot(sce, reduction = "tsne", group.by = "RNA_snn_res.1") +
ggtitle("louvain_1"))
p1_dim
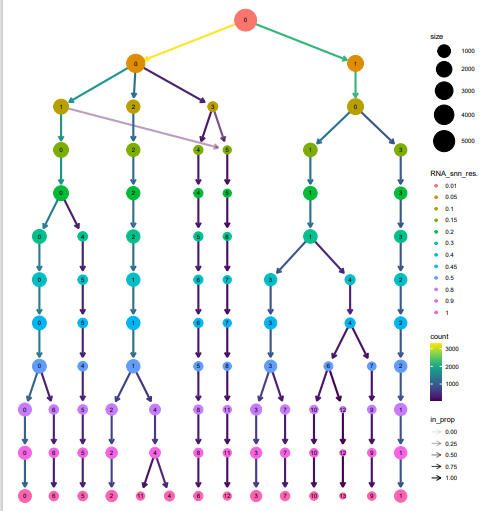
##(3)Дерево кластеризации
p2_tree = clustree(sce@meta.data, prefix = "RNA_snn_res.")
p2_tree
ggsave(plot = p2_tree, filename="Tree_diff_resolution.pdf",width = 10,height = 11)
##(4)Группа
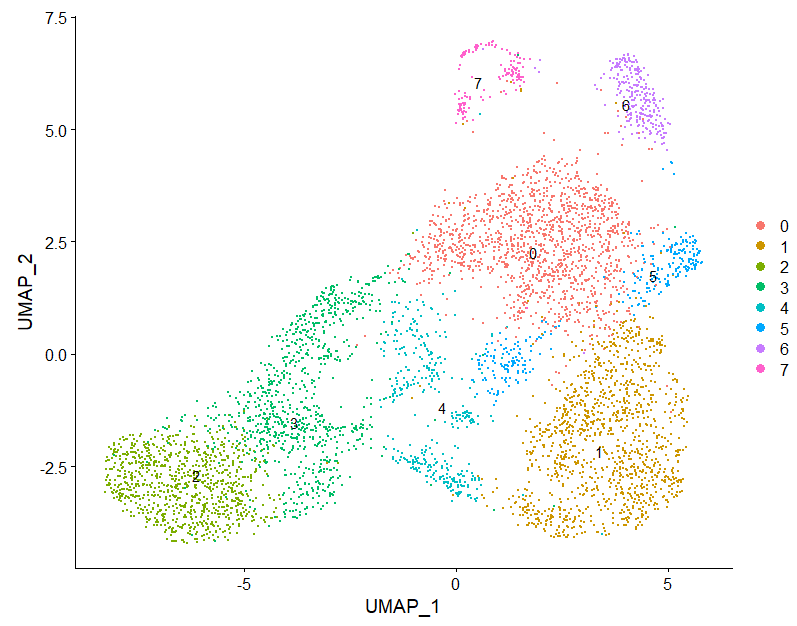
#Окончательное разрешение выбора кластеризации = 0,4, ячейки группируются в 7 кластеров.
sce1 <- FindClusters(sce, resolution = 0.4)
p1 <- DimPlot(sce1, reduction = "umap", label = TRUE, repel = TRUE)
p1
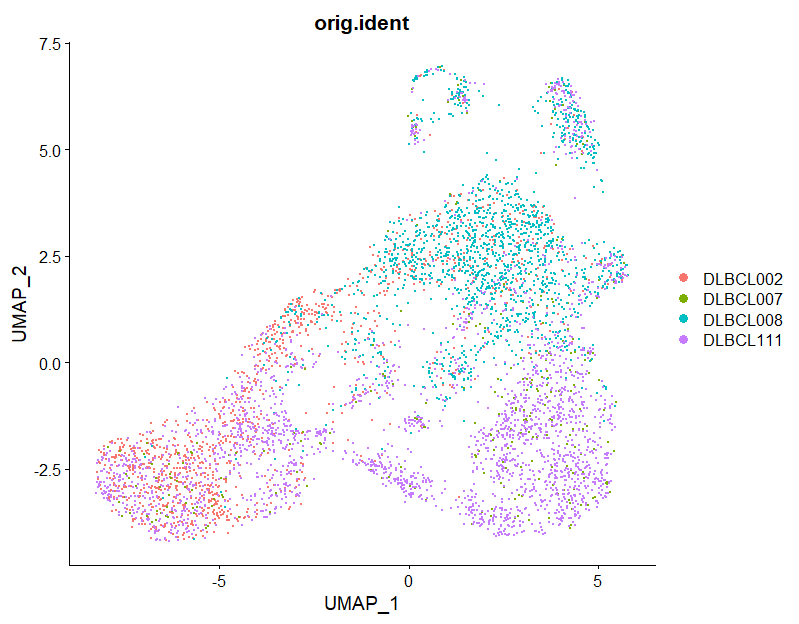
p2 <- DimPlot(sce1, reduction = "umap", group.by = "orig.ident")
p2
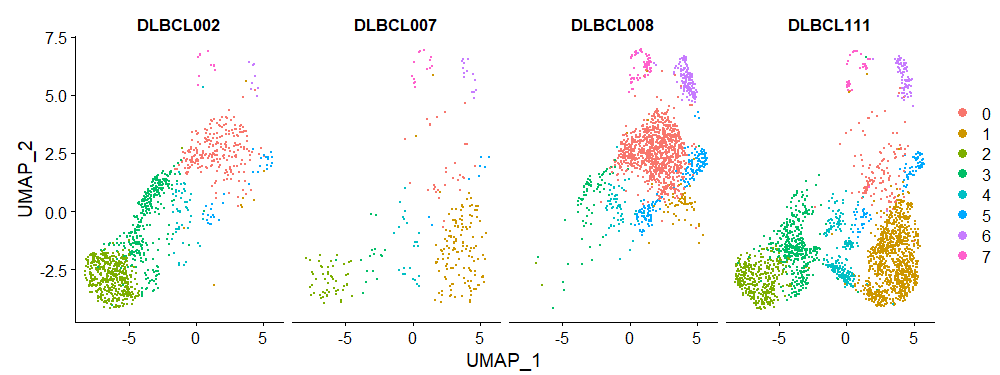
#Вы также можете наблюдать кластеризацию клеток в каждом образце индивидуально
p3=DimPlot(sce1, reduction = "umap", split.by = "orig.ident")
p3
#Извлечение маркерных генов каждого типа клеток
#использовать FindMarkers Команда, вы можете найти дифференциально экспрессируемые гены в каждом типе клеток по сравнению с другими категориями в качестве биологических маркерных генов для этого типа клеток. Параметр ident.1 задает категорию анализируемых клеток, а min.pct представляет собой долю числа экспрессий гена к общему количеству клеток этого типа.
#install.packages("metap")
library(metap)
#1_find all markers of cluster 1
cluster1.markers <- FindMarkers(sce, ident.1 = 6, min.pct = 0.25)
head(cluster1.markers, n = 5)
#использовать DoHeatmap Команда может предварительно просмотреть экспрессию гена-маркера.
pbmc.markers <- FindAllMarkers(sce, only.pos = TRUE, min.pct = 0.25)
#2_С кластером 6 Примеры обнаружения сохранившихся marker
DefaultAssay(sce1) <- "RNA"
nk.markers <- FindConservedMarkers(sce1, ident.1 = 6, grouping.var = "orig.ident", verbose = FALSE)
nk.markers #Выполните описанную выше операцию для каждого кластера, чтобы узнать сохранившиеся значения всех типов ячеек marker
View(nk.markers)
head(nk.markers, n = 9)
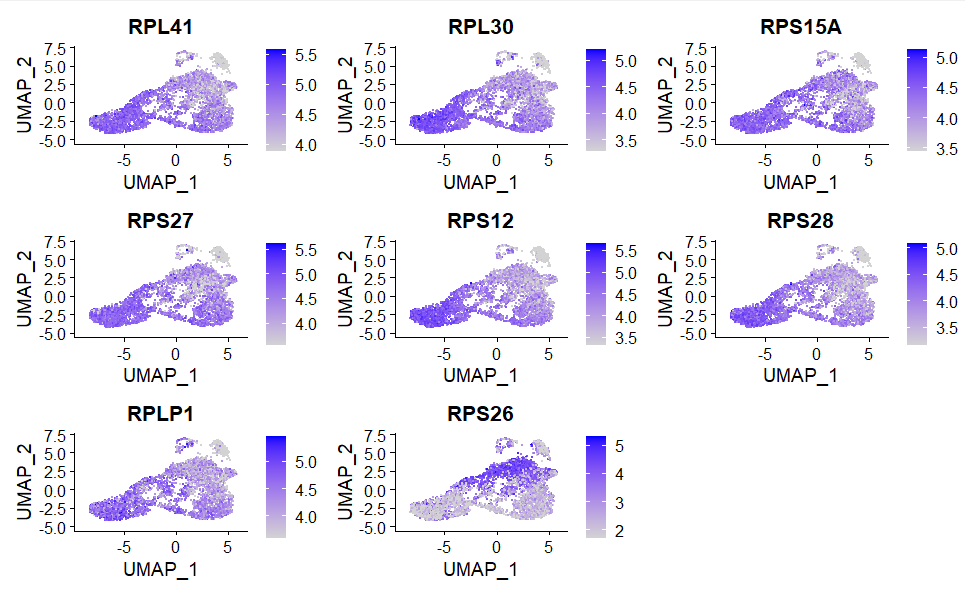
#Проверьте обилие маркеров в разных клетках
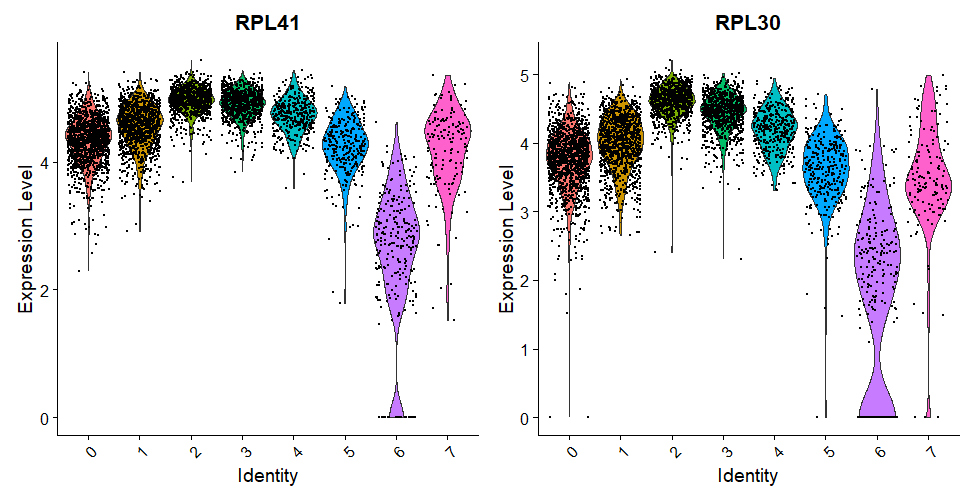
p4=FeaturePlot(sce1, features = c("RPL41", "RPL30", "RPS15A", "RPS27", "RPS12", "RPS28", "RPLP1",
"RPLP10", "RPS26"), min.cutoff = "q9")
ggsave(plot = p4, filename="FeaturePlot_conserved_marker.pdf",width = 10,height = 11)
#Исследуйте интересующие гены
#Seurat предлагает множество методов, которые позволяют нам удобно исследовать экспрессию интересующих генов в различных типах клеток.
VlnPlot(sce1, features = c("RPL41", "RPL30")) #Здесь мы случайным образом выбрали два гена
Дружеская акция в конце статьи
Настоятельно рекомендую порекомендовать его окружающимПостдок и молодой биолог ИП,Будьте более осведомлены о данных,Пусть их научные исследования выйдут на более высокий уровень:
- Лекция марафона по биоинформатике (купи одну, получи пять) , ваш вводный курс по биоинформатике
- Спустя 5 лет наше VIP-обучение в дереве биотехнологических навыков продолжает набирать студентов.
- 144 потока, 640 ГБ памяти, совместное использование сервера в течение одного года по-прежнему составляет всего 800
- Эксклюзивный облачный сервер биоинформатики, которого ждали уже давно
- Снова открыты заявки на стажеров по компиляции знаний Дерева навыков биотехнологии.
- Общий офис в Шэнсине в аренду


Учебное пособие по Jetpack Compose для начинающих, базовые элементы управления и макет

Код js веб-страницы, фон частицы, код спецэффектов

【новый! Суперподробное】Полное руководство по свойствам компонентов Figma.
🎉Обязательно к прочтению новичкам: полное руководство по написанию мини-программ WeChat с использованием программного обеспечения Cursor.
[Забавный проект Docker] VoceChat — еще одно приложение для мгновенного чата (IM)! Может быть встроен в любую веб-страницу!

Как реализовать переход по странице в HTML (html переходит на указанную страницу)

Как решить проблему зависания и низкой скорости при установке зависимостей с помощью npm. Существуют ли доступные источники npm, которые могут решить эту проблему?

Серия From Zero to Fun: Uni-App WeChat Payment Practice WeChat авторизует вход в систему и украшает страницу заказа, создает интерфейс заказа и инициирует запрос заказа

Серия uni-app: uni.navigateЧтобы передать скачок значения

Апплет WeChat настраивает верхнюю панель навигации и адаптируется к различным моделям.

JS-время конвертации

Обеспечьте бесперебойную работу ChromeDriver 125: советы по решению проблемы chromedriver.exe не найдены

Поле комментария, щелчок мышью, специальные эффекты, js-код
Объект массива перемещения объекта JS

Как открыть разрешение на позиционирование апплета WeChat_Как использовать WeChat для определения местонахождения друзей
Я даю вам два набора из 18 простых в использовании фонов холста Power BI, так что вам больше не придется возиться с цветами!

Получить текущее время в js_Как динамически отображать дату и время в js

Вам необходимо изучить сочетания клавиш vsCode для форматирования и организации кода, чтобы вам больше не приходилось настраивать формат вручную.

У ChatGPT большое обновление. Всего за 45 минут пресс-конференция показывает, что OpenAI сделал еще один шаг вперед.
Copilot облачной разработки — упрощение разработки

Микросборка xChatGPT с низким кодом, создание апплета чат-бота с искусственным интеллектом за пять шагов

CUDA Out of Memory: идеальное решение проблемы нехватки памяти CUDA
Анализ кластеризации отдельных ячеек, который должен освоить каждый&MarkerгенетическийВизуализация

vLLM: мощный инструмент для ускорения вывода ИИ

CodeGeeX: мощный инструмент генерации кода искусственного интеллекта, который можно использовать бесплатно в дополнение к второму пилоту.

Машинное обучение Реальный бой LightGBM + настройка параметров случайного поиска: точность 96,67%
Бесшовная интеграция, мгновенный интеллект [1]: платформа больших моделей Dify-LLM, интеграция без кодирования и встраивание в сторонние системы, более 42 тысяч звезд, чтобы стать свидетелями эксклюзивных интеллектуальных решений.
LM Studio для создания локальных больших моделей
Как определить количество слоев и нейронов скрытых слоев нейронной сети?
